A Translation-Based Method for Learned Molecular Descriptors
This is a Method paper that introduces Continuous and Data-Driven Descriptors (CDDD), a neural machine translation approach for learning fixed-size, continuous molecular representations. Rather than training an autoencoder to reconstruct SMILES strings, Winter et al. train an encoder-decoder model to translate between semantically equivalent but syntactically different molecular representations (e.g., randomized SMILES to canonical SMILES, or InChI to canonical SMILES). The bottleneck latent vector serves as a general-purpose molecular descriptor. Pretrained on approximately 72 million compounds from ZINC15 and PubChem, CDDD produces 512-dimensional descriptors that achieve competitive QSAR performance and significantly outperform all tested molecular fingerprints in ligand-based virtual screening.
Why Translation Instead of Reconstruction?
Molecular descriptors are central to cheminformatics. Traditional approaches rely on human-engineered fingerprints like ECFPs, which encode structural features as fixed-length bit vectors. While effective, these representations are constrained by predefined feature extraction rules.
Recent work applied deep neural networks directly to molecular graphs or SMILES strings to learn task-specific representations. However, these end-to-end approaches must learn features from scratch for each new dataset, making them prone to overfitting on the small bioactivity datasets typical in drug discovery.
Unsupervised approaches based on autoencoders (notably Gomez-Bombarelli et al.’s VAE and Xu et al.’s seq2seq model) offered a path toward general-purpose learned descriptors. These models reconstruct SMILES strings through an information bottleneck, forcing the latent space to capture molecular information. The concern with reconstruction, however, is that the model may focus on syntactic patterns of the string representation rather than the underlying chemical semantics. A model that memorizes SMILES syntax shortcuts can achieve low reconstruction error without truly encoding chemical meaning.
Winter et al. address this by drawing on the analogy to neural machine translation: a translator must understand the meaning of a sentence to produce a correct translation in another language. By training the model to translate between different molecular representations (which share chemical semantics but differ in syntax), the latent space is forced to capture the chemical information common to both representations, rather than representation-specific syntactic artifacts.
Translation as Semantic Compression
The core insight is that translating between two syntactically different but semantically equivalent representations forces the encoder to capture only the chemical meaning shared by both. The model architecture follows the standard encoder-decoder framework from neural machine translation.
The encoder reads a source molecular string (e.g., a randomized SMILES or InChI) and compresses it into a fixed-size latent vector. The decoder takes this latent vector and generates the target molecular string (canonical SMILES). The model is trained to minimize character-level cross-entropy between the decoder output and the target sequence.
Four translation tasks were evaluated:
- Randomized SMILES to canonical SMILES (best performing)
- InChI to canonical SMILES
- Canonical SMILES to canonical SMILES (autoencoding baseline)
- Canonical SMILES to InChI (failed to learn)
The final model uses an RNN encoder with 3 stacked GRU layers (512, 1024, and 2048 units). The concatenated cell states pass through a fully connected layer with tanh activation to produce a 512-dimensional latent vector. The decoder mirrors this architecture, initializing its GRU states from the latent vector via separate fully connected layers. Teacher forcing is used during training, and left-to-right beam search is used at inference.
An auxiliary property prediction network takes the latent vector as input and predicts nine molecular properties (logP, partial charges, valence electrons, H-bond donors/acceptors, Balaban’s J, molar refractivity, TPSA). This multi-task signal encourages the latent space to encode physically meaningful information. The full training objective combines the translation cross-entropy loss with the property prediction mean squared error:
$$\mathcal{L} = \mathcal{L}_{\text{translation}} + \mathcal{L}_{\text{properties}}$$
To ensure invariance to input SMILES representation at inference time, the model uses randomized SMILES as input half the time and canonical SMILES the other half during training. Input dropout (15% at the character level) and Gaussian noise (standard deviation 0.05) are applied for regularization.
QSAR Benchmarks, Virtual Screening, and Latent Space Exploration
Pretraining
The model was pretrained on approximately 72 million compounds from ZINC15 and PubChem (merged, deduplicated, filtered for organic molecules with MW 12-600, >3 heavy atoms, logP between -7 and 5). All evaluation compounds were removed from the pretraining set.
QSAR Experiments
Ten QSAR datasets were used, spanning classification (Ames mutagenicity, hERG inhibition, BBB penetration, BACE inhibition, bee toxicity) and regression (EGFR inhibition, Plasmodium falciparum inhibition, lipophilicity, aqueous solubility, melting point). Two datasets (Ames and lipophilicity) served as validation for architecture selection; the remaining eight were held out for final evaluation.
CDDD descriptors with an SVM were benchmarked against:
- Nine circular fingerprint variants (Morgan fingerprints, radius 1-3, folded to 512/1024/2048 bits) with RF, SVM, and GB
- Graph convolution models (DeepChem)
Both random-split and cluster-split (K-means on MACCS fingerprints, K=5) cross-validation were performed.
| Task | Split | CDDD + SVM | Best Fingerprint | Graph Conv |
|---|---|---|---|---|
| Ames (ROC-AUC) | Random | 0.89 | 0.89 (ecfc2, RF) | 0.88 |
| hERG (ROC-AUC) | Random | 0.86 | 0.85 (ecfc4, RF) | 0.86 |
| BBBP (ROC-AUC) | Random | 0.93 | 0.93 (ecfc2, RF) | 0.92 |
| BACE (ROC-AUC) | Random | 0.90 | 0.91 (ecfc2, RF) | 0.91 |
| Bee toxicity (ROC-AUC) | Random | 0.92 | 0.91 (ecfc6, RF) | 0.89 |
| Lipophilicity ($r^2$) | Random | 0.72 | 0.69 (ecfc2, SVM) | 0.73 |
| ESOL ($r^2$) | Random | 0.92 | 0.58 (ecfc6, SVM) | 0.86 |
| Melting point ($r^2$) | Random | 0.42 | 0.38 (ecfc2, SVM) | 0.39 |
CDDD descriptors showed competitive or better performance across all tasks. Notably, CDDD achieved substantially higher $r^2$ on aqueous solubility (0.92 vs. 0.58 for the best fingerprint). The authors emphasize that CDDD’s feature extraction was fixed based on two validation tasks, while baseline methods selected the best fingerprint/model combination per task, making the comparison conservative for CDDD.
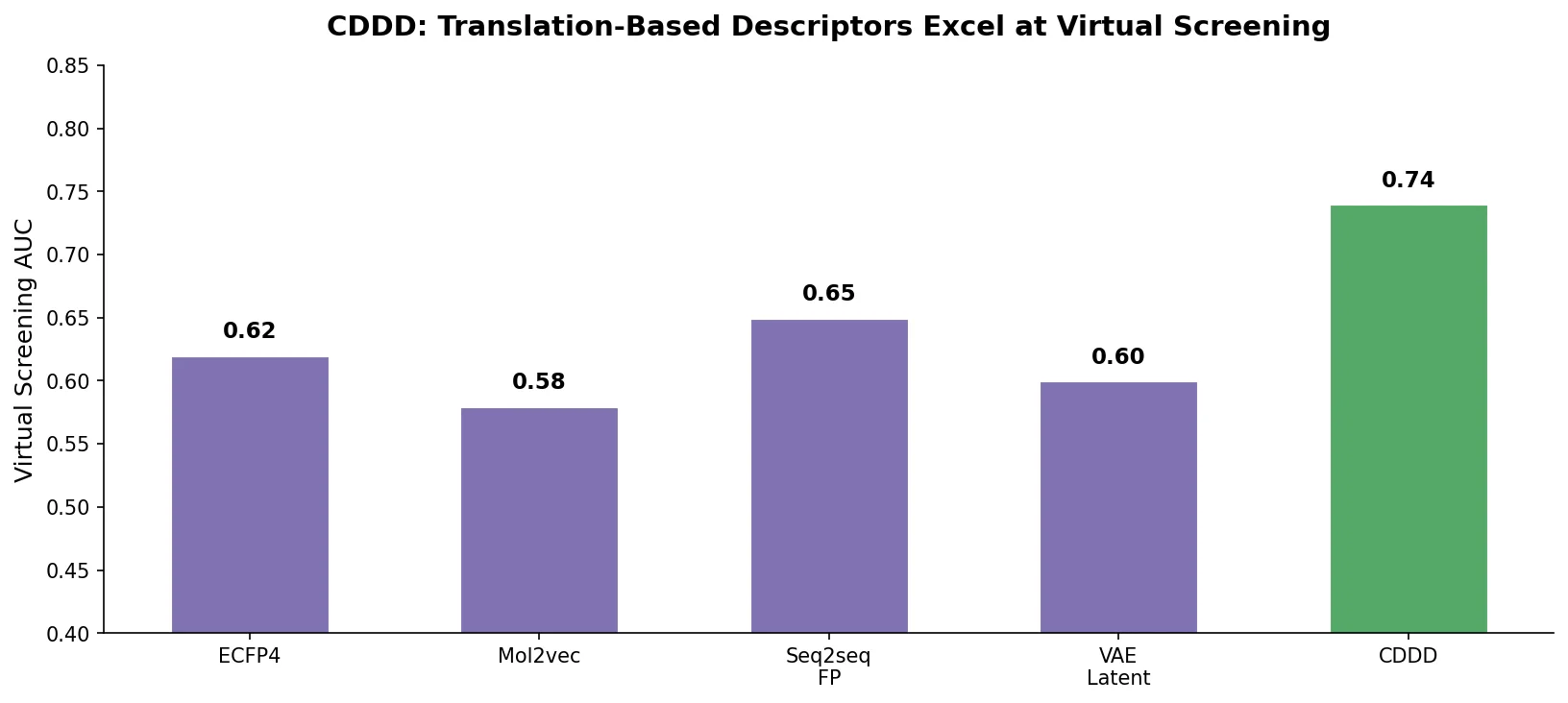
Virtual Screening
Ligand-based virtual screening experiments followed the Riniker et al. benchmarking protocol on 40 DUD targets and 17 MUV targets. Five active compounds were randomly selected per target, and remaining compounds were ranked by similarity (cosine similarity for CDDD, Tanimoto for fingerprints). This process was repeated 50 times per target.
| Database | CDDD (ROC-AUC) | Second Best | p-value (Wilcoxon) |
|---|---|---|---|
| DUD | 0.949 | 0.899 (laval) | $5 \times 10^{-38}$ |
| MUV | 0.679 | 0.677 (ap) | 0.04 |
CDDD significantly outperformed all 14 baseline fingerprints on both databases. The DUD improvement was particularly large (+5.0 ROC-AUC points over the next best). On MUV, which is designed to be harder, the advantage was smaller but still statistically significant. Importantly, while the best baseline fingerprint varied between DUD and MUV (laval vs. ap), CDDD ranked first on both, demonstrating consistent performance.
Latent Space Exploration
The continuous, reversible nature of CDDD enables chemical space navigation. Shifting a molecule’s embedding along the first principal component of the pretraining data correlates with molecular size (Spearman $r = 0.947$, $p = 0.00048$), while the second principal component correlates with polarity/logP ($r = -0.916$, $p = 0.00015$).
When shifting 1000 compounds along 100 random directions, the model maintained high valid SMILES generation rates (>97% for the top beam search output, >99% when considering the top 3 outputs). Euclidean distance in the descriptor space correlated smoothly with Tanimoto distance in fingerprint space, confirming that the latent space supports meaningful interpolation.
Consistent Learned Descriptors for Chemistry
CDDD demonstrated that translation between molecular representations produces more informative latent spaces than autoencoder reconstruction. The key findings are:
- Translation outperforms reconstruction: Models trained on translating between different representations consistently produced better downstream descriptors than autoencoding models, despite autoencoding being an easier task.
- Auxiliary property prediction helps: The additional classification task for molecular properties improved descriptor quality, particularly for physicochemical endpoints correlated with the predicted properties.
- Consistent performance: Unlike baseline methods where the best fingerprint varies by task, CDDD showed consistent performance across all QSAR and VS experiments.
- Smooth latent space: The continuous descriptor space supports meaningful interpolation and chemical space exploration with high valid SMILES rates.
The authors acknowledge several limitations. The InChI-to-SMILES translation worked but produced inferior descriptors compared to SMILES-to-SMILES, and SMILES-to-InChI translation failed entirely, likely due to InChI’s complex syntax (counting, arithmetic). The approach was only tested with string-based representations; translation between conceptually different representations (e.g., 3D structures) remains future work. The QSAR evaluation, while extensive, used relatively standard datasets, and the method’s advantage over graph convolution models was modest on tasks where end-to-end learning had sufficient data.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining | ZINC15 + PubChem (merged) | ~72M compounds | Filtered: organic, MW 12-600, >3 heavy atoms, logP -7 to 5 |
| Validation | Ames mutagenicity | 6,130 | Classification |
| Validation | Lipophilicity | 3,817 | Regression |
| Test | hERG, BBBP, BACE, bee toxicity | 188-3,440 | Classification |
| Test | EGFR, Plasmodium, ESOL, melting point | 184-4,451 | Regression |
| VS | DUD | 40 targets | Ligand-based virtual screening |
| VS | MUV | 17 targets | Maximum unbiased validation |
Algorithms
- Encoder: 3 stacked GRU layers (512, 1024, 2048 units) with tanh bottleneck to 512-dim latent space
- Decoder: Matching 3 stacked GRU layers, initialized from latent space
- Auxiliary classifier: 3 FC layers (512, 128, 9) predicting molecular properties
- Optimizer: Adam, initial LR $5 \times 10^{-4}$, decayed by 0.9 every 50,000 steps
- Batch size: 64 with bucketing by sequence length
- Input regularization: 15% character dropout + Gaussian noise (std 0.05)
- Beam search for decoding at inference
Models
| Artifact | Type | License | Notes |
|---|---|---|---|
| CDDD (GitHub) | Code + Model | MIT | Pretrained model and extraction code |
Evaluation
- QSAR: 5-fold random CV and 5-fold cluster CV (K-means on MACCS, K=5)
- Classification metric: ROC-AUC
- Regression metric: $r^2$
- VS: ROC-AUC averaged over 50 random active set selections per target
- Statistical test: Wilcoxon signed-rank test for VS comparisons
Hardware
- Framework: TensorFlow 1.4.1
- Fingerprint extraction on GPU is comparable in speed to RDKit on CPU
- SVM training on 512-dim CDDD descriptors takes seconds (vs. minutes for 2048-dim fingerprints)
- Graph convolution training: ~30 minutes per task on GPU
Paper Information
Citation: Winter, R., Montanari, F., Noe, F., & Clevert, D.-A. (2019). Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chemical Science, 10(6), 1692-1701. https://doi.org/10.1039/C8SC04175J
@article{winter2019learning,
title={Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations},
author={Winter, Robin and Montanari, Floriane and No{\'e}, Frank and Clevert, Djork-Arn{\'e}},
journal={Chemical Science},
volume={10},
number={6},
pages={1692--1701},
year={2019},
publisher={Royal Society of Chemistry},
doi={10.1039/C8SC04175J}
}