An Empirical Framework for Probing Chemical Understanding
This is an Empirical paper that introduces Augmented Molecular Retrieval (AMORE), a zero-shot evaluation framework for chemical language models (ChemLMs). The primary contribution is a method to assess whether ChemLMs have learned genuine molecular semantics or simply memorize textual patterns. Rather than relying on traditional NLP metrics like BLEU and ROUGE, AMORE tests whether a model’s embedding space treats chemically equivalent SMILES representations as similar. The authors evaluate 12 models across multiple architectures (encoder-only, encoder-decoder, decoder-only) on two datasets and five augmentation types, and extend the analysis to downstream MoleculeNet tasks.
Why Standard NLP Metrics Fail for Chemical Evaluation
Chemical language models are typically evaluated using text-based metrics from NLP (BLEU, ROUGE, METEOR) on tasks like molecule captioning. These metrics compare word overlap and sentence fluency but cannot detect whether a model truly understands molecular structure. A SMILES string like C(=O)O and its canonicalized or kekulized form represent the same molecule, yet text-based metrics would penalize valid reformulations. Embedding-based metrics like BERTScore are also insufficient because they were trained on general text, not chemical notation.
The core research question is direct: do evaluation metrics used on ChemLMs reflect actual chemical knowledge, or do the models simply imitate understanding by learning textual features? This question has practical consequences in pharmaceuticals and healthcare, where missteps in chemical reasoning carry serious risks.
Embedding-Based Retrieval as a Chemical Litmus Test
AMORE exploits a fundamental property of molecular representations: a single molecule can be written as multiple valid SMILES strings that are chemically identical. These serve as “total synonyms,” a concept without a true analogue in natural language.
The framework works in four steps:
- Take a set $X = (x_1, x_2, \ldots, x_n)$ of $n$ molecular representations.
- Apply a transformation $f$ to obtain augmented representations $X’ = (x’_1, x’_2, \ldots, x’_n)$, where $x’_i = f(x_i)$. The constraint is that $f$ must not change the underlying molecule.
- Obtain vectorized embeddings $e(x_i)$ and $e(x’_j)$ from the model for each original and augmented SMILES.
- Evaluate in a retrieval task: given $e(x_i)$, retrieve $e(x’_i)$ from the augmented set.
The evaluation metrics are top-$k$ accuracy (whether the correct augmented SMILES ranks at position $\leq k$) and Mean Reciprocal Rank (MRR). Retrieval uses FAISS for efficient nearest-neighbor search. The key insight is that if a model truly understands molecular structure, it should embed different SMILES representations of the same molecule close together.
Five SMILES Augmentation Types
The framework uses five identity-preserving augmentations, all executed through RDKit:
- Canonicalization: Transform SMILES to the standardized RDKit canonical form.
- Hydrogen addition: Explicitly add hydrogen atoms that are normally implied (e.g.,
Cbecomes[CH4]). This dramatically increases string length. - Kekulization: Convert aromatic ring notation to explicit alternating double bonds.
- Cycle renumbering: Replace ring-closure digit identifiers with random valid alternatives.
- Random atom order: Randomize the atom traversal order used to generate the SMILES string.
Twelve Models, Two Datasets, Five Augmentations
Models Evaluated
The authors test 12 publicly available Transformer-based models spanning three architecture families:
| Model | Domain | Parameters |
|---|---|---|
| Text+Chem T5-standard | Cross-modal | 220M |
| Text+Chem T5-augm | Cross-modal | 220M |
| MolT5-base | Cross-modal | 220M |
| MolT5-large | Cross-modal | 770M |
| SciFive | Text-only | 220M |
| PubChemDeBERTa | Chemical | 86M |
| ChemBERT-ChEMBL | Chemical | 6M |
| ChemBERTa | Chemical | 125M |
| BARTSmiles | Chemical | 400M |
| ZINC-RoBERTa | Chemical | 102M |
| nach0 | Chemical | 220M |
| ZINC-GPT | Chemical | 87M |
Datasets
- ChEBI-20 test set: ~3,300 molecule-description pairs, used for both AMORE retrieval and molecule captioning comparisons.
- Isomers (QM9 subset): 918 molecules that are all isomers of C9H12N2O, making retrieval harder because all molecules share the same molecular formula.
Key Results on ChEBI-20
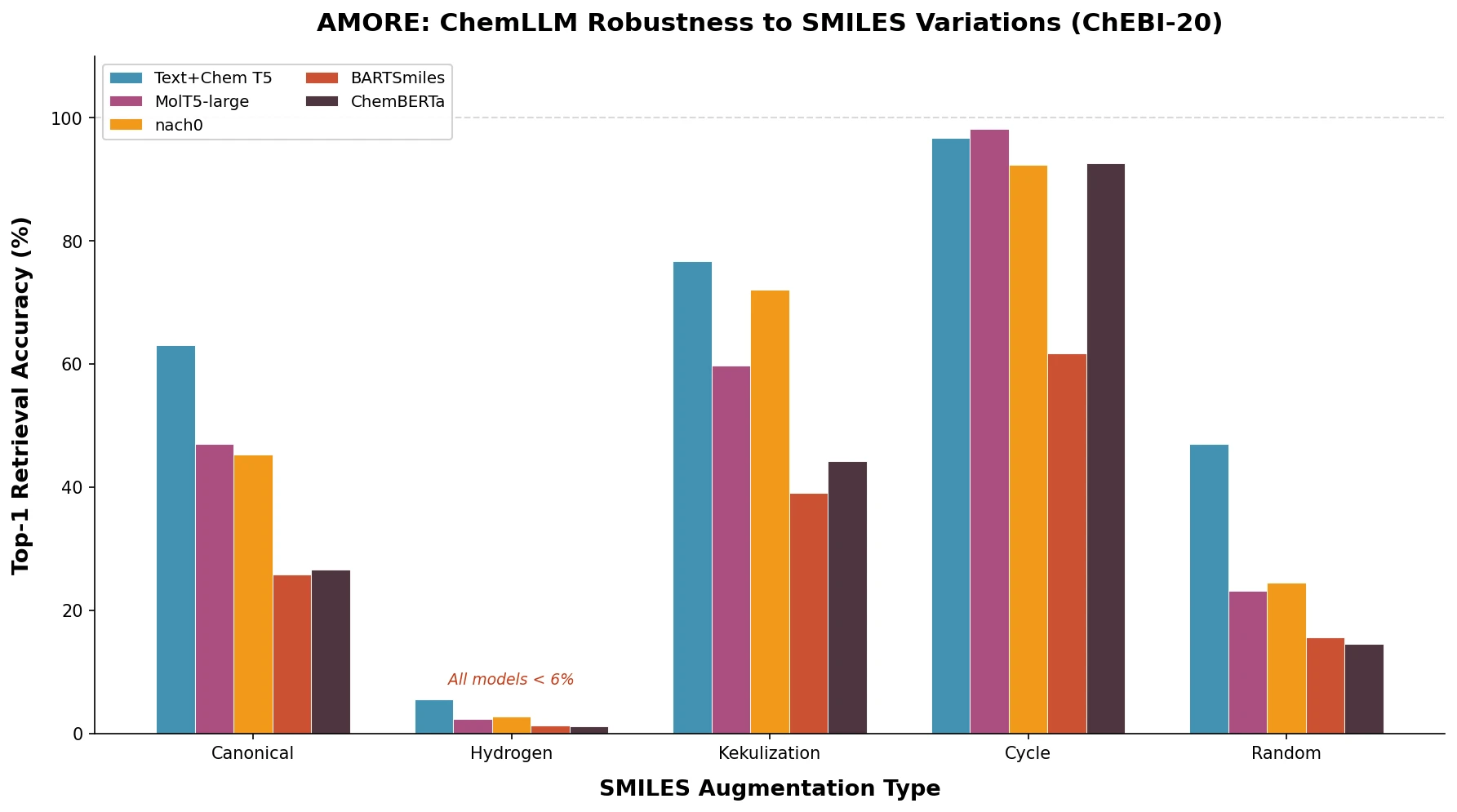
On the ChEBI-20 dataset (Table 2 from the paper), top-1 accuracy varies enormously by augmentation type. Cycle renumbering is easiest (up to 98.48% Acc@1 for SciFive), while hydrogen addition is hardest (no model exceeds 5.97% Acc@1).
For the cross-modal Text+Chem T5-standard model:
| Augmentation | Acc@1 | Acc@5 | MRR |
|---|---|---|---|
| Canonical | 63.03 | 82.76 | 72.4 |
| Hydrogen | 5.46 | 10.85 | 8.6 |
| Kekulization | 76.76 | 92.03 | 83.8 |
| Cycle | 96.70 | 99.82 | 98.2 |
| Random | 46.94 | 74.18 | 59.33 |
Key Results on Isomers
Performance drops substantially on the Isomers dataset, where all molecules share the same formula. The best Acc@1 for hydrogen augmentation is just 1.53% (MolT5-large). Even for the relatively easy cycle augmentation, top scores drop from the high 90s to the low 90s for most models, and some models (BARTSmiles: 41.83%) struggle considerably.
Downstream MoleculeNet Impact
The authors also fine-tuned models on original MoleculeNet training data and tested on augmented test sets across 9 tasks (regression, binary classification, multilabel classification). Results confirm that augmentations degrade downstream performance. For example, on ESOL regression, RMSE increased from 0.87 to 7.93 with hydrogen addition. Rankings computed using the Vote’n’Rank framework (using the Copeland rule) show that hydrogen augmentation is the only one that substantially reshuffles model rankings; other augmentations preserve the original ordering.
Correlation Between AMORE and Captioning Metrics
The differences in ROUGE/METEOR between original and augmented SMILES correlate with AMORE retrieval accuracy (Spearman correlation > 0.7 with p-value = 0.003 for Acc@1). This validates AMORE as a proxy for predicting how augmentations will affect generation quality, without requiring labeled captioning data.
Current ChemLMs Learn Syntax, Not Chemistry
The central finding is that existing ChemLMs are not robust to identity-preserving SMILES augmentations. Several specific conclusions emerge:
Hydrogen augmentation is catastrophic: All models fail (< 6% Acc@1 on ChEBI-20, < 2% on Isomers). The authors attribute this to the near-complete absence of explicit hydrogen in pretraining data, creating a distribution shift.
Cross-modal models outperform unimodal ones: Models trained on both text and SMILES (Text+Chem T5, MolT5) consistently achieve higher retrieval accuracy on four of five augmentations.
Augmentation difficulty follows a consistent order: For all models, hydrogen is hardest, followed by canonicalization, random ordering, kekulization, and cycle renumbering (easiest).
Layer-wise analysis reveals instability: Retrieval accuracy across Transformer layers is correlated across augmentation types, suggesting that representations degrade at the same layers regardless of augmentation.
Levenshtein distance partially explains difficulty: Hydrogen augmentation produces strings ~2x longer than originals (Levenshtein ratio of 1.49), but the low correlation between Levenshtein ratio and downstream metrics (ROUGE1 correlation of -0.05 for hydrogen) suggests string length alone does not explain the failure.
Limitations
The authors acknowledge several limitations. Only publicly available HuggingFace models were evaluated, excluding models like Chemformer and Molformer that lack HF checkpoints. The study focuses exclusively on SMILES sequences, not 3D molecular structures or other formats like SELFIES. The augmentation types, while representative, do not cover all possible identity transformations.
The authors suggest that AMORE could serve as a regularization tool during training, for example by using metric learning to encourage models to embed SMILES variants of the same molecule close together.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Retrieval evaluation | ChEBI-20 test set | 3,300 molecules | Standard benchmark for molecule captioning |
| Retrieval evaluation | Isomers (QM9 subset) | 918 molecules | All isomers of C9H12N2O |
| Downstream evaluation | MoleculeNet (9 tasks) | Varies | ESOL, Lipophilicity, FreeSolv, HIV, BBBP, BACE, Tox21, ToxCast, SIDER |
Algorithms
- SMILES augmentations via RDKit (canonicalization, hydrogen addition, kekulization, cycle renumbering, random atom ordering)
- Nearest-neighbor retrieval using FAISS with L2, cosine, inner product, and HNSW metrics
- Model ranking via Vote’n’Rank (Copeland rule) on MoleculeNet tasks
Models
All 12 evaluated models are publicly available on HuggingFace. No custom model training was performed for the AMORE retrieval experiments. MoleculeNet experiments used standard fine-tuning on original training splits.
Evaluation
| Metric | Description | Notes |
|---|---|---|
| Acc@1 | Top-1 retrieval accuracy | Primary AMORE metric |
| Acc@5 | Top-5 retrieval accuracy | Secondary AMORE metric |
| MRR | Mean Reciprocal Rank | Average rank of correct match |
| ROUGE-2 | Bigram overlap for captioning | Compared against AMORE |
| METEOR | MT evaluation metric for captioning | Compared against AMORE |
Hardware
Computational resources from HPC facilities at HSE University. Specific GPU types and training times are not reported.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| AMORE GitHub | Code | Not specified | Framework code and evaluation data |
Paper Information
Citation: Ganeeva, V., Khrabrov, K., Kadurin, A., & Tutubalina, E. (2025). Measuring Chemical LLM robustness to molecular representations: a SMILES variation-based framework. Journal of Cheminformatics, 17(1). https://doi.org/10.1186/s13321-025-01079-0
@article{ganeeva2025measuring,
title={Measuring Chemical LLM robustness to molecular representations: a SMILES variation-based framework},
author={Ganeeva, Veronika and Khrabrov, Kuzma and Kadurin, Artur and Tutubalina, Elena},
journal={Journal of Cheminformatics},
volume={17},
number={1},
year={2025},
publisher={Springer},
doi={10.1186/s13321-025-01079-0}
}