Paper Information
Citation: Flam-Shepherd, D., Zhu, K., & Aspuru-Guzik, A. (2022). Language models can learn complex molecular distributions. Nature Communications, 13, 3293.
Publication: Nature Communications, 2022
Additional Resources:
@article{flamshepherd2022language,
title={Language models can learn complex molecular distributions},
author={Flam-Shepherd, Daniel and Zhu, Kevin and Aspuru-Guzik, Al{\'a}n},
journal={Nature Communications},
volume={13},
pages={3293},
year={2022},
publisher={Nature Publishing Group},
doi={10.1038/s41467-022-30839-x}
}
RNN Language Models as Flexible Molecular Generators
This is an Empirical paper that investigates the capacity of simple recurrent neural network (RNN) language models to learn complex molecular distributions. The core finding is that LSTM-based models trained on SMILES (SM-RNN) or SELFIES (SF-RNN) string representations consistently outperform popular graph generative models (JTVAE, CGVAE) across three increasingly challenging generative modeling tasks. The paper positions language models as flexible, scalable alternatives to graph-based approaches for molecular generation.
Scaling Beyond Standard Benchmarks
Most molecular generative models are evaluated on relatively small, drug-like molecules from datasets like ZINC or MOSES. These standard benchmarks do not test whether models can handle larger, more structurally diverse molecules or distributions with complex shapes (multi-modal, heavy-tailed). This gap matters because there is increasing interest in larger, more complex molecules for therapeutics, including peptides and natural products.
Graph generative models like JTVAE and CGVAE impose structural constraints (tree decompositions, valency restrictions) that help with validity but limit their ability to scale. Language models, by contrast, only need to generate a single character sequence, making them inherently more flexible.
Three Challenging Generative Modeling Tasks
The paper introduces three benchmark tasks designed to stress-test generative models:
Task 1: Penalized LogP Distribution
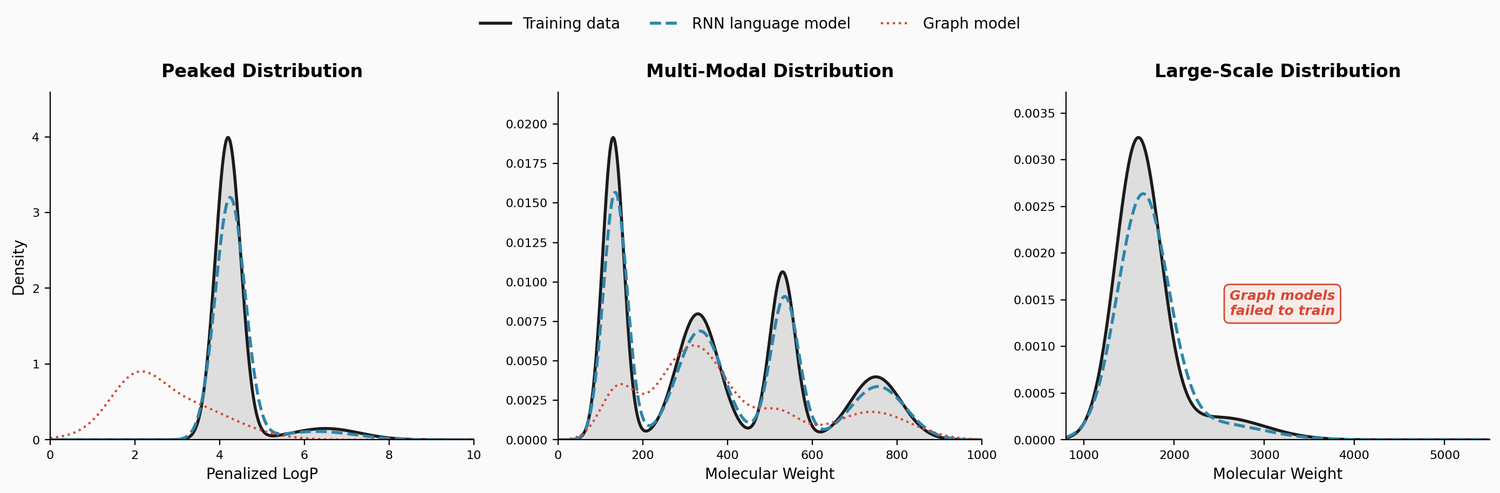
A dataset of approximately 160K molecules from ZINC15 with penalized LogP scores exceeding 4.0. The training distribution is sharply peaked around 4.0 to 4.5 with a subtle tail extending above 6.0. Molecules in the tail tend to have long carbon chains and fewer rings. The challenge is learning this skewed distribution rather than just finding individual high-scoring molecules.
Task 2: Multi-Modal Distribution
A composite dataset of approximately 200K molecules drawn from four sources with distinct molecular weight ranges:
- GDB13 (MW $\leq$ 185)
- ZINC (185 $\leq$ MW $\leq$ 425)
- Harvard Clean Energy Project (460 $\leq$ MW $\leq$ 600)
- POLYMERS (MW $>$ 600)
Models must learn to generate from all four modes simultaneously, each with very different molecular structures.
Task 3: Large-Scale Molecules
The largest molecules in PubChem with more than 100 heavy atoms, yielding approximately 300K molecules with molecular weights ranging from 1,250 to 5,000. These include small biomolecules, photovoltaics, peptides, and cyclic peptides. This task is particularly challenging because the SMILES/SELFIES strings are very long.
Evaluation by Distributional Fidelity
The evaluation framework focuses on how well a model learns the full training distribution rather than generating individual good molecules. The primary quantitative metric is the Wasserstein distance (earth mover’s distance) between molecular property distributions of generated and training molecules:
$$W(P, Q) = \inf_{\gamma \in \Gamma(P,Q)} \int | x - y | , d\gamma(x, y)$$
Properties evaluated include LogP, synthetic accessibility (SA), quantitative estimate of drug-likeness (QED), molecular weight (MW), Bertz complexity (BCT), and natural product likeness (NP). An oracle baseline is computed by measuring the Wasserstein distance between different random samples of the training data itself.
Standard metrics (validity, uniqueness, novelty) are also reported but are secondary to distributional fidelity.
Architecture: LSTM Language Models
The language models use standard LSTM architectures trained autoregressively on molecular strings. Two variants are compared:
- SM-RNN: Trained on canonical SMILES
- SF-RNN: Trained on SELFIES representations
Hyperparameters are tuned via random search over learning rate ($\in [0.0001, 0.001]$), hidden units ($\in [100, 1000]$), layers (1 to 5), and dropout ($\in [0.0, 0.5]$). Model selection uses a combination of standard metrics and Wasserstein distance rankings.
The graph model baselines include JTVAE (junction tree VAE) and CGVAE (constrained graph VAE), along with several additional baselines (MolGAN, GraphNVP, and others).
Results: Language Models Outperform Graph Models Across All Tasks
Penalized LogP
Both RNN models learn the sharp training distribution far better than graph models. The SM-RNN achieves the lowest Wasserstein distances across most properties. The graph models produce substantial out-of-distribution mass around penalized LogP scores of 1.75 to 2.25, failing to capture the peaked nature of the training distribution.
Critically, the RNNs also learn the subtle tail above penalized LogP of 6.0, generating molecules with long carbon chains and fewer rings that match the structural characteristics of high-scoring training molecules. CGVAE and JTVAE almost entirely miss this tail.
Multi-Modal Distribution
Both RNN models capture all four modes of the training distribution. JTVAE entirely misses the GDB13 mode and poorly learns the ZINC and CEP modes. CGVAE learns GDB13 but misses the CEP mode. The SM-RNN again achieves the best Wasserstein metrics.
Large-Scale Molecules
This is the most discriminating task. Both JTVAE and CGVAE completely fail to train on these large molecules. JTVAE’s tree decomposition produces a vocabulary of approximately 11,000 substructures, making training intractable. Only the RNN models succeed, with the SF-RNN achieving slightly better distributional match due to SELFIES guaranteeing 100% validity even for very long strings.
Both RNN models also learn the bimodal LogP structure within the large-molecule distribution and can generate molecules with substructures resembling peptides, including backbone chains and standard amino acid side chains.
Summary of Wasserstein Distance Results
| Task | Model | LogP | SA | QED | MW |
|---|---|---|---|---|---|
| LogP | SM-RNN | 0.095 | 0.031 | 0.007 | 3.3 |
| LogP | SF-RNN | 0.177 | 0.290 | 0.010 | 6.3 |
| LogP | JTVAE | 0.536 | 0.289 | 0.081 | 35.9 |
| LogP | CGVAE | 1.000 | 2.120 | 0.115 | 69.3 |
| Multi | SM-RNN | 0.081 | 0.025 | 0.006 | 5.5 |
| Multi | SF-RNN | 0.286 | 0.179 | 0.023 | 11.4 |
| Multi | JTVAE | 0.495 | 0.274 | 0.034 | 27.7 |
| Multi | CGVAE | 1.617 | 1.802 | 0.076 | 30.3 |
| Large | SM-RNN | 1.367 | 0.213 | 0.003 | 124.5 |
| Large | SF-RNN | 1.095 | 0.342 | 0.010 | 67.3 |
| Large | JTVAE | – | – | – | – |
| Large | CGVAE | – | – | – | – |
SMILES vs. SELFIES Trade-off
An interesting finding is that SMILES and SELFIES RNNs each have complementary strengths. The SF-RNN consistently achieves better standard metrics (validity, uniqueness, novelty) across all tasks, while the SM-RNN achieves better Wasserstein distance metrics. The authors suggest that the SELFIES grammar may reduce memorization of the training data, improving novelty but slightly hurting distributional fidelity.
Limitations
The authors acknowledge several limitations. Language models cannot account for molecular geometry or 3D information, which is important for many applications. The study evaluates distributional fidelity but does not test downstream utility for specific molecular design tasks (e.g., optimizing for a particular biological target). Additionally, while the graph models (JTVAE, CGVAE) are more interpretable, the language models operate as black boxes over string representations. The comparison is also limited to two specific graph model architectures, and more recent or specialized graph models may close the performance gap. Finally, trained model weights are only available upon request rather than being publicly released.
Reproducibility
| Artifact | Type | License | Notes |
|---|---|---|---|
| danielflamshep/genmoltasks | Dataset | Apache-2.0 | Processed training data and generated samples |
Data: Three custom datasets constructed from ZINC15, GDB13, Harvard Clean Energy Project, POLYMERS, and PubChem. Processed data available at the GitHub repository.
Code: LSTM networks implemented in PyTorch using the char-rnn code from the MOSES repository. Baselines use the official JTVAE and CGVAE implementations. No unified training script is provided in the repository.
Evaluation: Wasserstein distances computed using SciPy. Molecular properties computed using RDKit. 10K molecules generated from each model for evaluation.
Hyperparameters: Task-specific configurations reported. For example, the LogP task SM-RNN uses 2 hidden layers with 400 units, dropout of 0.2, and learning rate of 0.0001.
Hardware: Models were trained using the Canada Computing Systems (Compute Canada). Specific GPU types and training times are not reported.