Paper Information
Citation: Eckmann, P., Sun, K., Zhao, B., Feng, M., Gilson, M. K., & Yu, R. (2022). LIMO: Latent Inceptionism for Targeted Molecule Generation. Proceedings of the 39th International Conference on Machine Learning (ICML 2022), PMLR 162, 5777.
Publication: ICML 2022
Additional Resources:
@inproceedings{eckmann2022limo,
title={LIMO: Latent Inceptionism for Targeted Molecule Generation},
author={Eckmann, Peter and Sun, Kunyang and Zhao, Bo and Feng, Mudong and Gilson, Michael K and Yu, Rose},
booktitle={International Conference on Machine Learning},
pages={5777},
year={2022},
organization={PMLR}
}
Gradient-Based Reverse Optimization in Molecular Latent Space
This is a Method paper that introduces LIMO, a framework for generating molecules with desired properties using gradient-based optimization on a VAE latent space. The key innovation is a stacked architecture where a property predictor operates on the decoded molecular representation rather than directly on the latent space, combined with an inceptionism-like technique that backpropagates through the frozen decoder and predictor to optimize the latent code. This approach is 6-8x faster than RL baselines and 12x faster than sampling-based approaches while producing molecules with higher binding affinities.
Slow Property Optimization in Existing Methods
Generating molecules with high binding affinity to target proteins is a central goal of early drug discovery, but existing computational approaches are slow when optimizing for properties that are expensive to evaluate (such as docking-based binding affinity). RL-based methods require many calls to the property function during training. Sampling-based approaches like MARS need hundreds of iterations. Latent optimization methods that predict properties directly from the latent space suffer from poor prediction accuracy because the mapping from latent space to molecular properties is difficult to learn.
The LIMO Framework
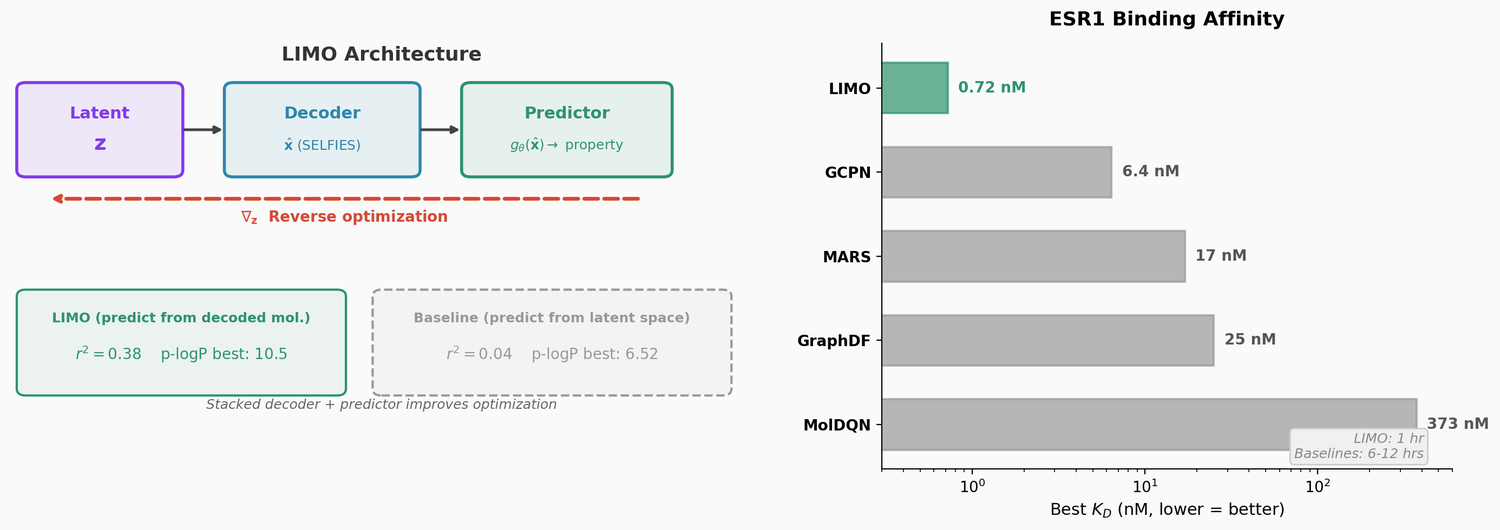
LIMO consists of three components: a VAE for learning a molecular latent space, a property predictor with a novel stacked architecture, and a gradient-based reverse optimization procedure.
SELFIES-Based VAE
The VAE encodes molecules represented as SELFIES strings into a 1024-dimensional latent space $\mathbf{z} \in \mathbb{R}^m$ and decodes to probability distributions over SELFIES symbols. Since all SELFIES strings correspond to valid molecules, this guarantees 100% chemical validity. The output molecule is obtained by taking the argmax at each position:
$$\hat{x}_i = s_{d_i^*}, \quad d_i^* = \operatorname{argmax}_{d} \{y_{i,1}, \ldots, y_{i,d}\}$$
The VAE uses fully-connected layers (not recurrent), with a 64-dimensional embedding layer, four batch-normalized linear layers (2000-dimensional first layer, 1000-dimensional for the rest) with ReLU activation, and is trained with ELBO loss (0.9 weight on reconstruction, 0.1 on KL divergence).
Stacked Property Predictor
The critical architectural choice: the property predictor $g_\theta$ takes the decoded molecular representation $\hat{\mathbf{x}}$ as input rather than the latent code $\mathbf{z}$. The predictor is trained after the VAE is frozen by minimizing MSE on VAE-generated molecules:
$$\ell_0(\theta) = \left\| g_\theta\left(f_{\text{dec}}(\mathbf{z})\right) - \pi\left(f_{\text{dec}}(\mathbf{z})\right) \right\|^2$$
where $\pi$ is the ground-truth property function. This stacking improves prediction accuracy from $r^2 = 0.04$ (predicting from $\mathbf{z}$) to $r^2 = 0.38$ (predicting from $\hat{\mathbf{x}}$) on an unseen test set. The improvement comes because the mapping from molecular space to property is easier to learn than the mapping from latent space to property.
Reverse Optimization (Inceptionism)
After training, the decoder and predictor weights are frozen and $\mathbf{z}$ becomes the trainable parameter. For multiple properties with weights $(w_1, \ldots, w_k)$, the optimization minimizes:
$$\ell_1(\mathbf{z}) = -\sum_{i=1}^{k} w_i \cdot g^i\left(f_{\text{dec}}(\mathbf{z})\right)$$
Since both the decoder and predictor are neural networks, gradients flow through the entire chain, enabling efficient optimization with Adam. This is analogous to the “inceptionism” (DeepDream) technique from computer vision, where network inputs are optimized to maximize specific outputs.
Substructure-Constrained Optimization
For lead optimization, LIMO can fix a molecular substructure during optimization by adding a regularization term:
$$\ell_2(\mathbf{z}) = \lambda \sum_{i=1}^{n} \sum_{j=1}^{d} \left(M_{i,j} \cdot \left(f_{\text{dec}}(\mathbf{z})_{i,j} - (\hat{\mathbf{x}}_{\text{start}})_{i,j}\right)\right)^2$$
where $M$ is a binary mask specifying which SELFIES positions must remain unchanged and $\lambda = 1000$. This capability is enabled by the intermediate decoded representation, which most VAE-based methods lack.
Experiments and Results
Benchmark Tasks (QED and Penalized LogP)
LIMO achieves competitive results with deep generative and RL-based models in 1 hour, compared to 8-24 hours for baselines. Top QED score: 0.947 (maximum possible: 0.948). Top penalized LogP: 10.5 (among length-limited models, comparable to MolDQN’s 11.8).
The ablation study (“LIMO on z”) confirms the stacked predictor architecture: predicting from $\hat{\mathbf{x}}$ yields top p-logP of 10.5 versus 6.52 when predicting directly from $\mathbf{z}$.
Binding Affinity Maximization
The primary contribution. LIMO generates molecules with substantially higher computed binding affinities (lower $K_D$) than baselines against two protein targets:
| Method | ESR1 best $K_D$ (nM) | ACAA1 best $K_D$ (nM) | Time (hrs) |
|---|---|---|---|
| GCPN | 6.4 | 75 | 6 |
| MolDQN | 373 | 240 | 6 |
| MARS | 17 | 163 | 6 |
| GraphDF | 25 | 370 | 12 |
| LIMO | 0.72 | 37 | 1 |
For ESR1, LIMO’s best molecule has a $K_D$ of 0.72 nM from docking, nearly 10x better than the next method (GCPN at 6.4 nM). When corroborated with more rigorous absolute binding free energy (ABFE) calculations, one LIMO compound achieved a predicted $K_D$ of $6 \times 10^{-14}$ M (0.00006 nM), far exceeding the affinities of approved drugs tamoxifen ($K_D$ = 1.5 nM) and raloxifene ($K_D$ = 0.03 nM).
Multi-Objective Optimization
Single-objective optimization produces molecules with high affinity but problematic structures (polyenes, large rings). Multi-objective optimization simultaneously targeting binding affinity, QED ($>$ 0.4), and SA ($<$ 5.5) produces drug-like, synthesizable molecules that still have nanomolar binding affinities. Generated molecules satisfy Lipinski’s rule of 5 with zero PAINS alerts.
Limitations
The LIMO property predictor achieves only moderate prediction accuracy ($r^2$ = 0.38), meaning the optimization relies on gradient direction being correct rather than absolute predictions being accurate. AutoDock-GPU docking scores do not correlate well with the more accurate ABFE results, a known limitation of docking. The fully-connected VAE architecture limits the molecular diversity compared to recurrent or attention-based alternatives (LSTM decoder produced max QED of only 0.3). The greedy fine-tuning step (replacing carbons with heteroatoms) is a heuristic rather than a learned procedure.
Reproducibility
| Artifact | Type | License | Notes |
|---|---|---|---|
| Rose-STL-Lab/LIMO | Code | UC San Diego Custom (non-commercial) | Full training, optimization, and evaluation code |
Data: ZINC250k dataset for optimization tasks. MOSES dataset for random generation evaluation. Binding affinities computed with AutoDock-GPU.
Hardware: Two GTX 1080 Ti GPUs (one for PyTorch, one for AutoDock-GPU), 4 CPU cores, 32 GB memory.
Training: VAE trained for 18 epochs with learning rate 0.0001. Property predictor uses 3 layers of 1000 units, trained for 5 epochs. Reverse optimization uses learning rate 0.1 for 10 epochs.
Targets: Human estrogen receptor (ESR1, PDB 1ERR) and human peroxisomal acetyl-CoA acyl transferase 1 (ACAA1, PDB 2IIK).