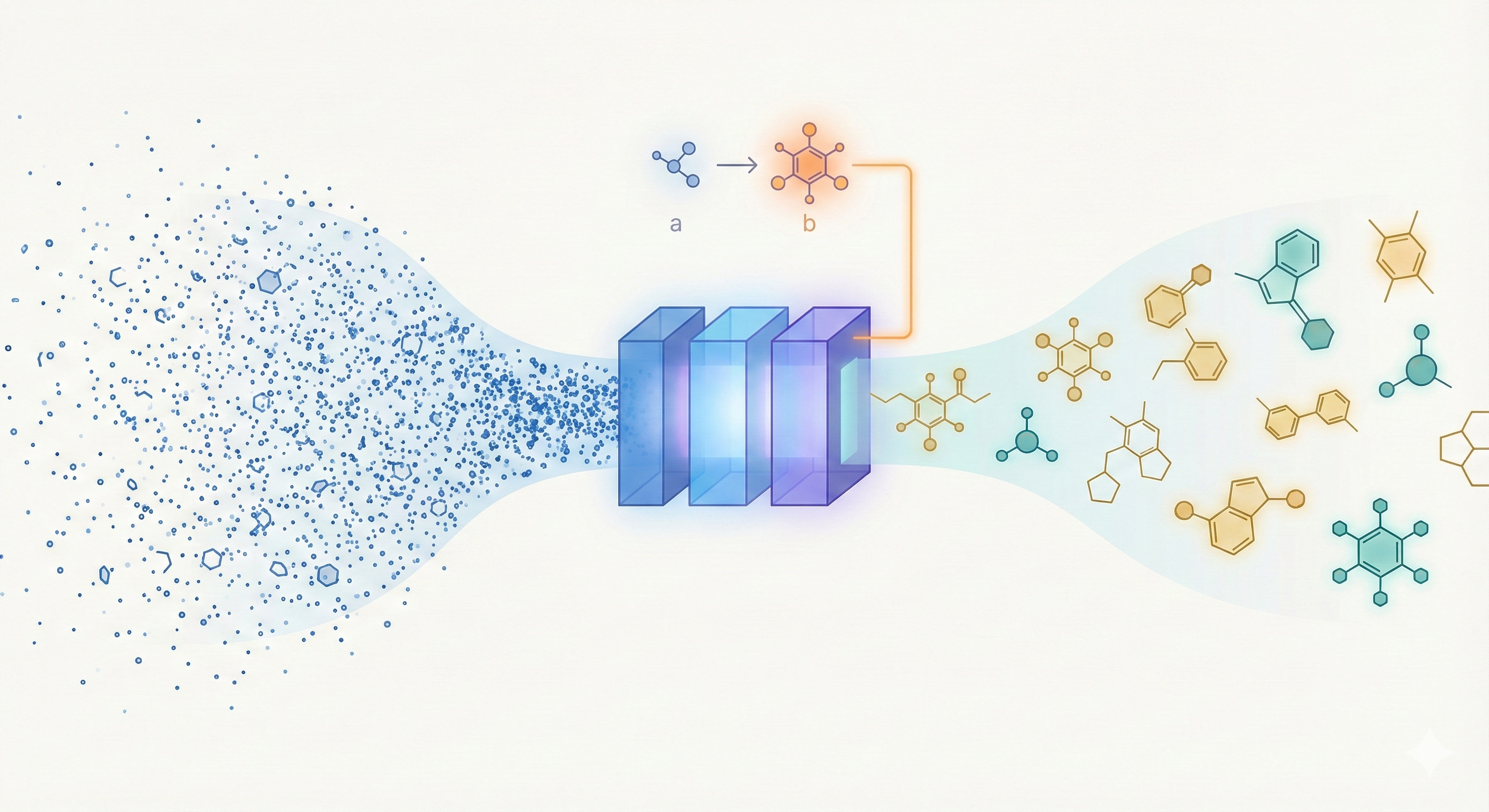
Contribution and Taxonomic Focus
This is primarily a Methodological paper, as it proposes a specific neural architecture (GP-MoLFormer) and a novel fine-tuning algorithm (Pair-tuning) for molecular generation. It validates these contributions against standard baselines (e.g., JT-VAE, MolGen-7b).
It also contains a secondary Theoretical contribution by establishing an empirical scaling law that relates inference compute (generation size) to the novelty of the generated molecules.
Motivation: Data Scale and Prompt-Based Optimization
While large language models (LLMs) have transformed text generation, the impact of training data scale and memorization on molecular generative models remains under-explored. Specifically, there is a need to understand how training on billion-scale datasets affects the novelty of generated molecules and whether biases in public databases (like ZINC and PubChem) perpetuate memorization. Furthermore, existing optimization methods often require computationally expensive property predictors or reinforcement learning loops; there is a practical need for more efficient “prompt-based” optimization techniques.
Core Innovations: Architecture and Pair-Tuning
- Architecture: The application of a linear-attention transformer decoder with Rotary Positional Embeddings (RoPE) to generative chemistry, allowing for efficient training on 1.1 billion SMILES.
- Pair-Tuning: A novel, parameter-efficient fine-tuning method that uses property-ordered molecular pairs to learn “soft prompts” for optimization without updating the base model weights.
- Scaling Analysis: An extensive empirical investigation mapping the trade-off between inference compute (up to 10B generations) and chemical novelty, fitting an exponential decay curve that demonstrates how novelty saturates as generation volume grows.
Experimental Methodology and Downstream Tasks
The authors evaluated GP-MoLFormer on three distinct tasks, though the comparisons highlight the difficulty of evaluating foundation models against classical baselines:
- De Novo Generation: Comparing validity, uniqueness, and novelty against baselines (CharRNN, VAE, LIMO, MolGen-7b) on a held-out test set. Notably, this is an unequal comparison; most baselines were trained on the 1.6M molecule MOSES dataset, whereas GP-MoLFormer uses up to 1.1B molecules, meaning performance gains are heavily driven by data scale.
- Scaffold-Constrained Decoration: Generating molecules from DRD2 active binder scaffolds and measuring the hit rate of active compounds against specialized scaffold decorators.
- Property-Guided Optimization: Using Pair-tuning to optimize for Drug-likeness (QED), Penalized logP, and DRD2 binding activity, comparing the results to graph-based and reinforcement learning benchmarks.
Additionally, they performed a Scaling Study:
- Comparing models trained on raw (1.1B) vs. de-duplicated (650M) data.
- Generating up to 10 billion molecules to fit empirical scaling laws for novelty.
Key Findings and Scaling Laws
- Scale Driven Performance: GP-MoLFormer achieves high internal diversity and validity on generation metrics. However, its baseline novelty percentage (~32%) is considerably lower than classical models. The authors attribute this to the massive training scale forcing the model to heavily prioritize matching real-world molecule frequencies over pure exploration. GP-MoLFormer’s advantage in generation metrics over LLM-baselines like MolGen-7b likely stems heavily from its 10x larger training dataset rather than fundamental architectural superiority.
- Pair-Tuning Efficacy: The proposed pair-tuning method effectively optimizes properties (e.g., improving DRD2 activity scores) without requiring full model fine-tuning or external reward loops. While successful, the text-based generation yields ~94.5% validity during optimization, which lags behind graph and SELFIES-based baselines that guarantee 100% structural validity.
- Memorization vs. Novelty: Training on de-duplicated data (GP-MoLFormer-UNIQ) yields higher novelty (approx. 5-8% higher) than training on raw data, confirming that duplication bias in public databases leads directly to memorization.
- Inference Scaling Law: Novelty decays exponentially with generation size ($y = ae^{-bx}$), yet the model maintains generative capability (~16.7% novelty) even after generating an unprecedented 10 billion molecules.
Reproducibility Details
Data
- Sources: A combination of PubChem (111M SMILES) and ZINC (1B SMILES) databases. Downloading and pre-training instructions are located in the repository’s
data/README.md. - Preprocessing:
- All SMILES were canonicalized using RDKit (no isomeric information).
- GP-MoLFormer (Base): Trained on the full 1.1B dataset (includes duplicates).
- GP-MoLFormer-UNIQ: Trained on a de-duplicated subset of 650M SMILES.
- Tokenization: Uses the tokenizer from Schwaller et al. (2019) with a vocabulary size of 2,362 tokens.
- Filtering: Sequences restricted to a maximum length of 202 tokens.
Algorithms
Pair-Tuning (Algorithm 1):
- Objective: Learn task-specific soft prompts $\phi_T$ to maximize the conditional probability of target molecule $b$ given a seed molecule $a$, where pair $(a, b)$ satisfies the property condition $b > a$. The base model parameters $\theta$ remain frozen.
- Prompt Structure: Autoregressive training optimizes the continuous embeddings of $n$ enhancement tokens against the cross-entropy loss of the target sequence: $$ \mathcal{L}(\phi_T) = - \sum_{i=1}^{|b|} \log P_{\theta}(b_i | \phi_T, a, b_{<i}) $$
- Hyperparameters: Trained for 1,000 epochs with a batch size of 35 and a fixed learning rate of $3 \times 10^{-2}$.
- Inference: The learned prompt $\phi_T$ and seed molecule $a$ are prepended as context, and candidates are sampled autoregressively until a termination token is produced.
Models
- Availability: The model trained on deduplicated data (GP-MoLFormer-UNIQ) is publicly available on Hugging Face. The full 1.1B base model is not explicitly hosted. The source code repository includes a disclosure that IBM will not maintain the code going forward.
- Architecture: Transformer decoder (~47M parameters: 12 layers, 12 heads, hidden size 768).
- Attention Mechanism: Combines Linear Attention (Generalized Random Feature map, $\phi$) with Rotary Positional Embeddings (RoPE). To avoid the quadratic complexity of standard attention while maintaining relative positional awareness, RoPE is applied to queries ($Q$) and keys ($K$) prior to the random feature mapping: $$ \text{Attention}(Q, K, V) = \frac{\sum_{n=1}^N \langle \phi(R_m q_m), \phi(R_n k_n) \rangle v_n}{\sum_{n=1}^N \langle \phi(R_m q_m), \phi(R_n k_n) \rangle} $$
- Inference Speed: ~3ms per forward pass on a single A100 GPU.
Evaluation
- Generation Quality Metrics: Validity, Uniqueness, Novelty (MOSES suite), Fréchet ChemNet Distance (FCD), Scaffold similarity (Scaf), and Similarity to Nearest Neighbor (SNN).
- MoLFormer-Based Metrics: The authors introduce Fréchet MoLFormer Distance (FMD) and MoLFormer-space IntDiv2 to measure distributional similarity using their own pre-trained continuous embeddings instead of standard fingerprints.
- Optimization Metrics: Penalized logP (calculated as $\text{logP} - \text{SA} - \text{max}(\text{maxrings}(size) - 6, 0)$), Drug-likeness (QED), and DRD2 activity scores.
- Scaling Metrics: Empirical fit for novelty decay: $y = ae^{-bx}$.
Hardware
- Compute: 16 x NVIDIA A100 (80 GB) GPUs across 2 nodes connected via EDR Infiniband.
- Training Time:
- GP-MoLFormer (1.1B data): ~115 hours total (28.75 hours/epoch for 4 epochs).
- GP-MoLFormer-UNIQ (650M data): ~80 hours total.
- Hyperparameters: Used a batch size of 1,600 molecules per GPU with a fixed learning rate of $1.6 \times 10^{-4}$ (scaled up to $8\times$ factor as GPUs increased).
- Optimization: Used distributed data-parallel training and adaptive bucketing by sequence length to handle scale.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| GP-MoLFormer (GitHub) | Code | Apache 2.0 | Official implementation; IBM will not maintain going forward |
| GP-MoLFormer-Uniq (Hugging Face) | Model | Apache 2.0 | Pre-trained on 650M de-duplicated SMILES |
The full 1.1B base model weights are not publicly hosted. The training data (PubChem and ZINC) is publicly available, and instructions for downloading and pre-processing are in the repository’s data/README.md.
Paper Information
Citation: Ross, J., Belgodere, B., Hoffman, S. C., Chenthamarakshan, V., Navratil, J., Mroueh, Y., & Das, P. (2025). GP-MoLFormer: A Foundation Model For Molecular Generation. Digital Discovery (2025). https://doi.org/10.1039/D5DD00122F
Publication: Digital Discovery (2025)
@article{ross2025gpmolformer,
title={GP-MoLFormer: a foundation model for molecular generation},
author={Ross, Jerret and Belgodere, Brian and Hoffman, Samuel C and Chenthamarakshan, Vijil and Navratil, Jiri and Mroueh, Youssef and Das, Payel},
journal={Digital Discovery},
year={2025},
publisher={Royal Society of Chemistry},
doi={10.1039/D5DD00122F}
}