Paper Information
Citation: van Tilborg, D., Alenicheva, A., & Grisoni, F. (2022). Exposing the Limitations of Molecular Machine Learning with Activity Cliffs. Journal of Chemical Information and Modeling, 62(23), 5938-5951. https://doi.org/10.1021/acs.jcim.2c01073
Publication: Journal of Chemical Information and Modeling 2022
Additional Resources:
A Benchmark for Activity Cliff Prediction
This is a Systematization paper ($\Psi_{\text{Systematization}}$) with a significant Resource component ($\Psi_{\text{Resource}}$).
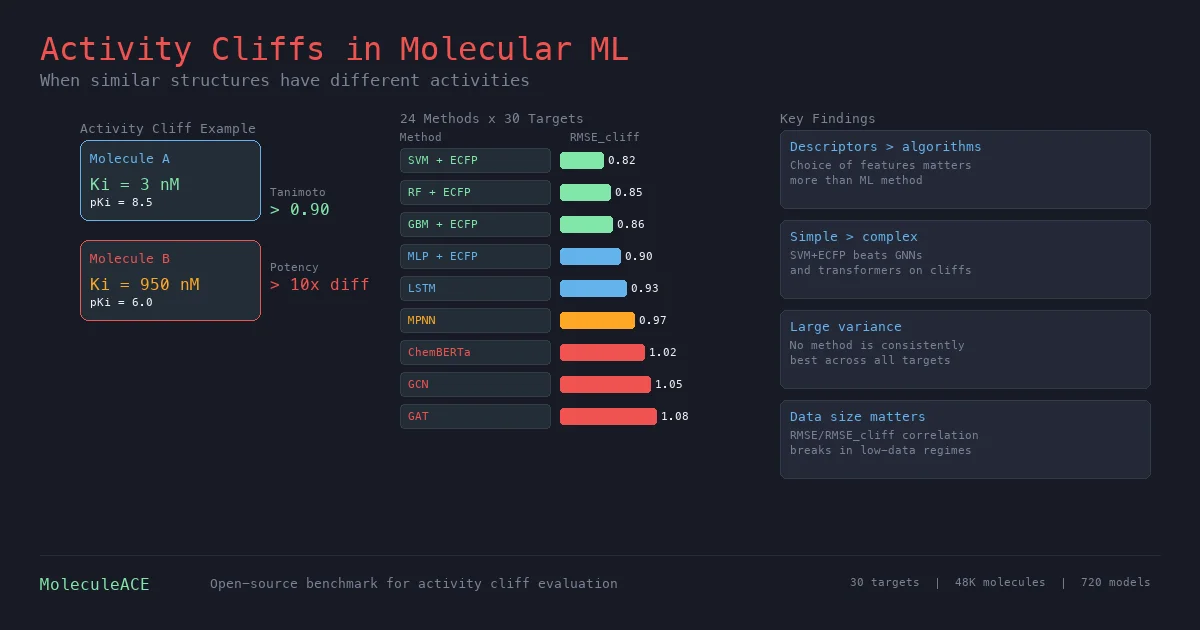
The paper systematically benchmarks 24 machine learning and deep learning approaches on their ability to predict bioactivity for activity cliff compounds: pairs of structurally similar molecules that exhibit large differences in potency. These cases violate the similarity principle (similar structure implies similar activity) and represent a practical failure mode for molecular property prediction in drug discovery. The authors release MoleculeACE, an open-source benchmarking platform for evaluating ML models on activity cliffs.
Activity Cliffs as a Blind Spot in Molecular ML
The similarity principle underpins most molecular ML: structurally similar compounds should have similar properties. Activity cliffs are the exceptions, where small structural changes cause large potency shifts (e.g., a single substituent change causing a 10x difference in $K_i$).
Despite their importance for hit-to-lead optimization, activity cliffs have received limited attention in ML benchmarking. Standard metrics like RMSE computed over entire test sets can mask poor predictions on cliff compounds. A model might achieve low overall error while systematically mispredicting these edge cases, which are precisely the molecules that matter most for medicinal chemistry applications.
The authors identify 7-52% of compounds as activity cliff molecules across their 30 target datasets, showing this is not a rare phenomenon.
Defining and Detecting Activity Cliffs
The authors use three complementary similarity metrics to identify activity cliffs:
- Substructure similarity: Tanimoto coefficient on extended connectivity fingerprints (ECFPs), capturing shared radial substructures
- Scaffold similarity: Tanimoto coefficient on ECFPs computed from molecular graph frameworks, detecting core/decoration differences
- SMILES similarity: Levenshtein distance on canonical SMILES strings, capturing character-level insertions, deletions, and translocations
Pairs with $\geq 90%$ similarity on any one of the three metrics and $> 10\times$ difference in bioactivity ($K_i$ or $\text{EC}_{50}$) are classified as activity cliff pairs. This union-based approach (rather than requiring agreement across all metrics) captures different types of structural relationships relevant to medicinal chemistry.
24 Methods Across 30 Drug Targets
The benchmark evaluates 16 traditional ML configurations (4 algorithms $\times$ 4 descriptor types) and 8 deep learning approaches across 30 curated ChEMBL v29 datasets (48,707 total molecules).
Traditional ML algorithms: KNN, RF, GBM, SVM, each combined with ECFPs, MACCS keys, WHIM descriptors, or physicochemical properties.
Deep learning methods: MPNN, GCN, GAT, Attentive FP (graph-based), plus LSTM, CNN, Transformer/ChemBERTa (SMILES-based), and an MLP on ECFPs.
Performance is measured with both standard RMSE and a dedicated $\text{RMSE}_{\text{cliff}}$ computed only on activity cliff compounds in the test set:
$$ \text{RMSE}_{\text{cliff}} = \sqrt{\frac{\sum_{j=1}^{n_c} (\hat{y}_j - y_j)^2}{n_c}} $$
Key results:
- Molecular descriptors matter more than algorithms: The choice of descriptor (ECFPs vs. MACCS vs. WHIM vs. physicochemical) had a larger impact on $\text{RMSE}_{\text{cliff}}$ than the choice of ML algorithm ($p < 0.05$, Wilcoxon rank-sum test with Benjamini-Hochberg correction).
- SVM + ECFPs wins on average: The best overall method for activity cliff prediction, though the difference from RF + ECFPs or GBM + ECFPs was not statistically significant.
- Deep learning underperforms: All graph and SMILES-based deep learning methods performed worse than a simple MLP on ECFPs. Among deep learning, LSTM with transfer learning (pretrained on 36K molecules) was the best, outperforming the ChemBERTa transformer pretrained on 10M compounds.
- Large case-by-case variation: $\text{RMSE}_{\text{cliff}}$ ranged from 0.62 to 1.60 log units across datasets, with no method consistently best. Deep learning methods showed the highest variance across targets.
Simple Descriptors Beat Complex Architectures on Cliffs
The core finding is that activity cliffs expose a gap in learned molecular representations. Despite graph neural networks and transformers being able to learn directly from molecular structure, they fail to capture the subtle structural differences that drive activity cliffs.
Key observations:
- RMSE and $\text{RMSE}_{\text{cliff}}$ correlate ($r = 0.81$ on average), so optimizing overall error usually helps with cliffs too. But this correlation breaks down for some targets (e.g., CLK4), where methods with similar RMSE can have very different $\text{RMSE}_{\text{cliff}}$.
- Training set size matters for the RMSE/$\text{RMSE}_{\text{cliff}}$ correlation: Datasets with $> 1000$ training molecules show $r > 0.80$ between the two metrics. In low-data regimes, the correlation weakens, making dedicated cliff evaluation more important.
- No relationship between % cliff compounds and model performance, and no target-family-specific effects were found.
- Transfer learning helped SMILES models (LSTM) but not graph models: Self-supervised pretraining strategies (context prediction, infomax, edge prediction, masking) did not improve GNN performance, consistent with findings from other studies.
The MoleculeACE platform provides standardized data curation, activity cliff detection, and cliff-specific evaluation, enabling researchers to assess new methods against this benchmark.
Reproducibility Details
Data
| Purpose | Source | Size | Notes |
|---|---|---|---|
| Benchmarking | ChEMBL v29 | 48,707 molecules (35,632 unique) across 30 targets | Curated for duplicates, salts, outliers |
| Smallest dataset | JAK1 | 615 molecules | 7% activity cliffs |
| Largest dataset | DRD3 | 3,657 molecules | 39% activity cliffs |
Algorithms
- Activity cliff detection: Pairwise similarity $\geq 0.9$ (Tanimoto on ECFPs, scaffold ECFPs, or Levenshtein on SMILES) with $> 10\times$ potency difference
- Splitting: Spectral clustering on ECFPs (5 clusters), 80/20 stratified split preserving cliff proportion
- Hyperparameter optimization: Bayesian optimization with Gaussian process, max 50 combinations, 5-fold cross-validation
- SMILES augmentation: 10-fold for all SMILES-based methods
- Transfer learning: LSTM pretrained on 36,281 merged training molecules (next-character prediction); ChemBERTa pretrained on 10M PubChem compounds
Models
- Traditional ML: KNN, RF, GBM, SVM (scikit-learn v1.0.2)
- Descriptors: ECFPs (1024-bit, radius 2), MACCS keys (166-bit), WHIM (114 descriptors), physicochemical (11 properties)
- GNNs: MPNN, GCN, GAT, AFP (PyTorch Geometric v2.0.4), with graph multiset transformer pooling
- SMILES models: LSTM (4 layers, 5.8M params), 1D CNN, ChemBERTa transformer
- Total models trained: 720 (24 methods $\times$ 30 targets)
Evaluation
| Metric | Scope | Details |
|---|---|---|
| RMSE | All test molecules | Standard root-mean-square error on $\text{pK}_i$ / $\text{pEC}_{50}$ |
| $\text{RMSE}_{\text{cliff}}$ | Activity cliff compounds only | RMSE restricted to cliff molecules in test set |
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| MoleculeACE | Code + Data | MIT | Benchmark platform with all 30 curated datasets |
| Curated datasets | Data | MIT | Processed ChEMBL bioactivity data |
Citation
@article{vantilborg2022activity,
title={Exposing the Limitations of Molecular Machine Learning with Activity Cliffs},
author={van Tilborg, Derek and Alenicheva, Alisa and Grisoni, Francesca},
journal={Journal of Chemical Information and Modeling},
volume={62},
number={23},
pages={5938--5951},
year={2022},
publisher={American Chemical Society},
doi={10.1021/acs.jcim.2c01073}
}