Paper Information
Citation: Cieplinski, T., Danel, T., Podlewska, S., & Jastrzebski, S. (2023). Generative Models Should at Least Be Able to Design Molecules That Dock Well: A New Benchmark. Journal of Chemical Information and Modeling, 63(11), 3238-3247. https://doi.org/10.1021/acs.jcim.2c01355
Publication: Journal of Chemical Information and Modeling 2023
Additional Resources:
A Docking-Based Benchmark for De Novo Drug Design
This is a Resource paper. Its primary contribution is a standardized benchmark for evaluating generative models in de novo drug design. Rather than introducing a new generative method, the paper provides a reusable evaluation framework built around molecular docking, a widely used computational proxy for predicting protein-ligand binding. The benchmark uses SMINA (a fork of AutoDock Vina) to score generated molecules against eight protein targets, offering a more realistic evaluation than commonly used proxy metrics like logP or QED.
Why Existing Benchmarks Fall Short
De novo drug design methods are typically evaluated using simple proxy tasks that do not reflect the complexity of real drug discovery. The octanol-water partition coefficient (logP) can be trivially optimized by producing unrealistic molecules. The QED drug-likeness score suffers from the same issue. Neural network-based bioactivity predictors are similarly exploitable.
As Coley et al. (2020) note: “The current evaluations for generative models do not reflect the complexity of real discovery problems.”
More realistic evaluation approaches exist in adjacent domains (photovoltaics, excitation energies), where physical calculations are used to both train and evaluate models. Yet de novo drug design has largely relied on the same simplistic proxies. This gap between proxy task performance and real-world utility motivates the development of a docking-based benchmark that, while still a proxy, captures more of the structural complexity involved in protein-ligand interactions.
Benchmark Design: SMINA Docking with the Vinardo Scoring Function
The benchmark is defined by three components: (1) docking software that computes a ligand’s pose in the binding site, (2) a scoring function that evaluates the pose, and (3) a training set of compounds with precomputed docking scores.
The concrete instantiation uses SMINA v. 2017.11.9 with the Vinardo scoring function:
$$S = -0.045 \cdot G + 0.8 \cdot R - 0.035 \cdot H - 0.6 \cdot B$$
where $S$ is the docking score, $G$ is the gauss term, $R$ is repulsion, $H$ is the hydrophobic term, and $B$ is the non-directional hydrogen bond term. The gauss and repulsion terms measure steric interactions between the ligand and the protein, while the hydrophobic and hydrogen bond terms capture favorable non-covalent contacts.
The benchmark includes three task variants:
- Docking Score Function: Optimize the full Vinardo docking score (lower is better).
- Repulsion: Minimize only the repulsion component, defined as:
$$ R(a_1, a_2) = \begin{cases} d(a_1, a_2)^2 & d(a_1, a_2) < 0 \\ 0 & \text{otherwise} \end{cases} $$
where $d(a_1, a_2)$ is the inter-atomic distance minus the sum of van der Waals radii.
- Hydrogen Bonding: Maximize the hydrogen bond term:
$$ B(a_1, a_2) = \begin{cases} 0 & (a_1, a_2) \text{ do not form H-bond} \\ 1 & d(a_1, a_2) < -0.6 \\ 0 & d(a_1, a_2) \geq 0 \\ \frac{d(a_1, a_2)}{-0.6} & \text{otherwise} \end{cases} $$
Scores are averaged over the top 5 binding poses for stability. Generated compounds are filtered by Lipinski’s Rule of Five and a minimum molecular weight of 100. Each model must generate 250 unique molecules per target.
Training data comes from ChEMBL, covering eight drug targets: 5-HT1B, 5-HT2B, ACM2, CYP2D6, ADRB1, MOR, A2A, and D2. Dataset sizes range from 1,082 (ADRB1) to 10,225 (MOR) molecules.
Experimental Evaluation of Three Generative Models
Models Tested
Three popular generative models were evaluated:
- CVAE (Chemical Variational Autoencoder): A VAE operating on SMILES strings.
- GVAE (Grammar Variational Autoencoder): Extends CVAE by enforcing grammatical correctness of generated SMILES.
- REINVENT: A recurrent neural network trained first on ChEMBL in a supervised manner, then fine-tuned with reinforcement learning using docking scores as rewards.
For CVAE and GVAE, molecules are generated by sampling from the latent space and taking 50 gradient steps to optimize an MLP that predicts the docking score. For REINVENT, a random forest model predicts docking scores from ECFP fingerprints, and the reward combines this prediction with the QED score.
Baselines
Two baselines provide context:
- Training set: The top 50%, 10%, and 1% of docking scores from the ChEMBL training set.
- ZINC subset: A random sample of ~9.2 million drug-like molecules from ZINC, with the same percentile breakdowns.
Diversity is measured as the mean Tanimoto distance (using 1024-bit ECFP with radius 2) between all pairs of generated molecules.
Key Results
| Task | Model | 5-HT1B Score | 5-HT1B Diversity |
|---|---|---|---|
| Docking Score | CVAE | -4.647 | 0.907 |
| Docking Score | GVAE | -4.955 | 0.901 |
| Docking Score | REINVENT | -9.774 | 0.506 |
| Docking Score | ZINC (10%) | -9.894 | 0.862 |
| Docking Score | ZINC (1%) | -10.496 | 0.861 |
| Docking Score | Train (10%) | -10.837 | 0.749 |
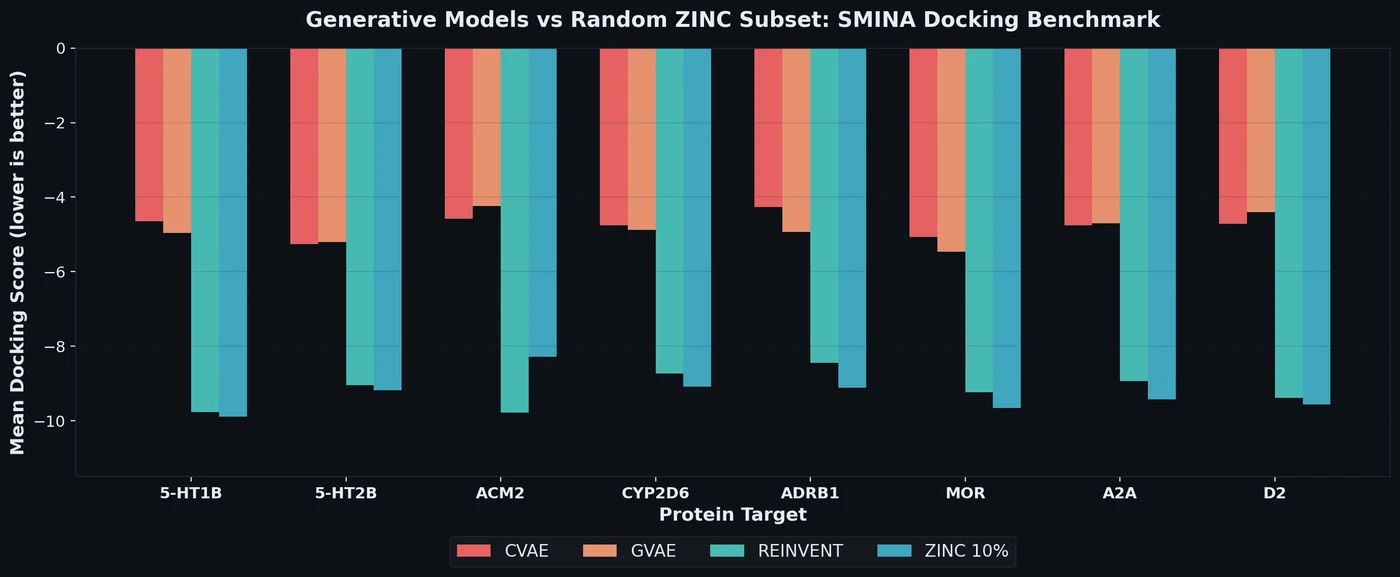
On the full docking score task, CVAE and GVAE fail to match even the mean ZINC docking score. REINVENT performs substantially better (e.g., -9.774 on 5-HT1B) but still falls short of the top 10% ZINC scores (-9.894) in most cases. The exception is ACM2, where REINVENT’s score (-9.775) exceeds the ZINC 10% threshold (-8.282).
On the repulsion task, all three models fail to outperform the top 10% ZINC scores. On the hydrogen bonding task (the easiest), GVAE and REINVENT nearly match the top 1% ZINC scores, suggesting that optimizing individual scoring components is more tractable than the full docking score.
A consistent finding across all experiments is that REINVENT generates substantially less diverse molecules than the training set (e.g., 0.506 vs. 0.787 mean Tanimoto distance on 5-HT1B). The t-SNE visualizations show generated molecules clustering in a single dense region, separate from the training data, regardless of optimization target.
The paper also notes a moderately strong correlation between docking scores and molecular weight or the number of rotatable bonds. Generated compounds achieve better docking scores at the same molecular weight after optimization, suggesting the models learn some structural preferences rather than simply exploiting molecular size.
Limitations of Current Generative Models for Drug Design
The main finding is negative: popular generative models for de novo drug design struggle to generate molecules that dock well when trained on realistically sized datasets (1,000 to 10,000 compounds). Even the best-performing model (REINVENT) generally cannot outperform the top 10% of a random ZINC subset on the full docking score task.
The authors acknowledge several limitations:
- Docking is itself a proxy: The SMINA docking score is only an approximation of true binding affinity. The fact that even this simpler proxy is challenging should raise concerns about these models’ readiness for real drug discovery pipelines.
- Limited model selection: Only three models were tested (CVAE, GVAE, REINVENT). The authors note that CVAE and GVAE were not designed for small training sets, and REINVENT may not represent the state of the art in all respects.
- ML-based scoring surrogate: All models use an ML model (MLP or random forest) to predict docking scores during generation, rather than running SMINA directly. This introduces an additional approximation layer.
- No similarity constraints: The benchmark does not impose constraints on the distance between generated and training molecules. A trivial baseline is to simply return the training set.
On a more positive note, the tested models perform well on the simplest subtask (hydrogen bonding), suggesting that optimizing docking scores from limited data is attainable but challenging. The benchmark has already been adopted by other groups, notably Nigam et al. (2021) for evaluating their JANUS genetic algorithm.
Future directions include adding similarity constraints, extending to additional protein targets, and using the benchmark to evaluate newer structure-based generative models that employ equivariant neural networks.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training/Evaluation | ChEMBL (8 targets) | 1,082-10,225 molecules per target | 90/10 train/test split |
| Baseline | ZINC 15 subset | ~9.2M drug-like molecules | In-stock, standard reactivity, drug-like |
| Protein structures | Protein Data Bank | 8 structures | Cleaned with Schrodinger modeling package |
Algorithms
- CVAE/GVAE: Fine-tuned 5 epochs on target data, then 50 gradient steps in latent space to optimize MLP-predicted score
- REINVENT: Pretrained on ChEMBL, fine-tuned with RL; reward = random forest prediction * QED score
- All docking performed with SMINA v. 2017.11.9 using Vinardo scoring function in score_only mode
- Scores averaged over top 5 binding poses
- Filtering: Lipinski Rule of Five, minimum molecular weight 100
Evaluation
| Metric | Description | Notes |
|---|---|---|
| Mean docking score | Average over 250 generated molecules | Lower is better for docking score and repulsion |
| Diversity | Mean Tanimoto distance (ECFP, r=2) | Higher is more diverse |
| ZINC percentile baselines | Top 50%, 10%, 1% from random ZINC subset | Task considered “solved” if generated score exceeds ZINC 1% |
Hardware
Not specified in the paper.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| smina-docking-benchmark | Code | MIT | Benchmark code, data, evaluation notebooks |
Citation
@article{cieplinski2023generative,
title={Generative Models Should at Least Be Able to Design Molecules That Dock Well: A New Benchmark},
author={Cieplinski, Tobiasz and Danel, Tomasz and Podlewska, Sabina and Jastrzebski, Stanislaw},
journal={Journal of Chemical Information and Modeling},
volume={63},
number={11},
pages={3238--3247},
year={2023},
publisher={American Chemical Society},
doi={10.1021/acs.jcim.2c01355}
}