Evaluating Chemical Foundation Models Through Surface Roughness
This is a Systematization paper that introduces a metric reformulation (ROGI-XD) and uses it to evaluate whether pretrained chemical models (PCMs) learn representations that produce smoother quantitative structure-property relationship (QSPR) surfaces than simple baselines. The key finding is negative: pretrained representations are no smoother than molecular fingerprints or descriptors, offering a principled explanation for their inconsistent performance on property prediction benchmarks.
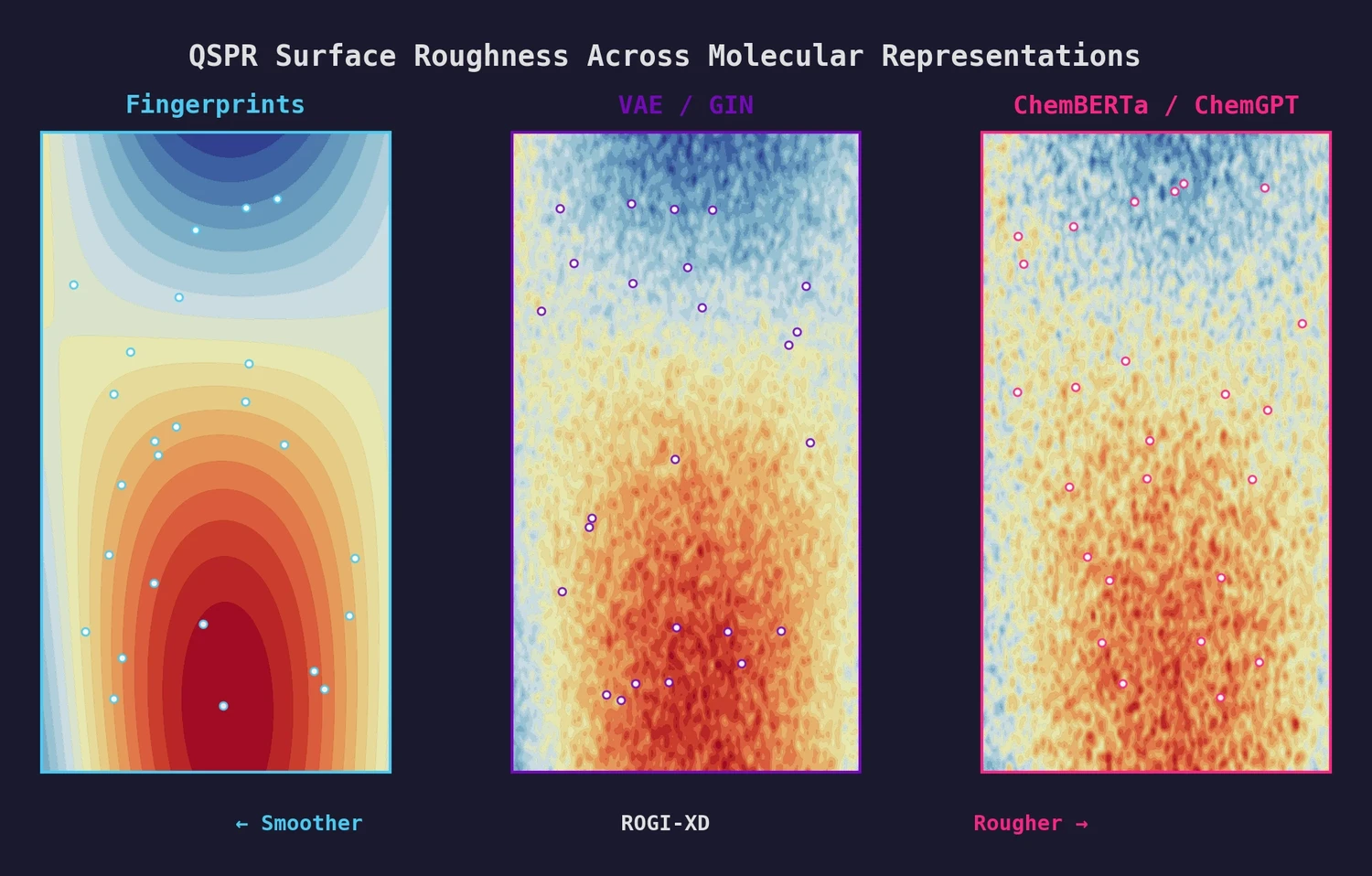
The Smoothness Gap in Chemical Foundation Models
Chemical foundation models like ChemBERTa, ChemGPT, and graph-based pretrained networks promise to learn meaningful molecular representations from large unlabeled datasets via self-supervised learning. However, empirical benchmarks consistently show mixed results: these learned representations sometimes match and sometimes underperform simple baselines like Morgan fingerprints or RDKit descriptors.
Prior work by Deng et al. demonstrated that a random forest trained on 2048-bit Morgan fingerprints was competitive with, or superior to, pretrained models like MolBERT and GROVER on MoleculeNet and opioid bioactivity tasks. The authors sought to explain this pattern through the lens of QSPR surface roughness: if pretrained representations do not produce smoother mappings from molecular structure to property, they cannot consistently outperform baselines.
ROGI-XD: A Dimensionality-Independent Roughness Metric
The original ROuGhness Index (ROGI) captures global surface roughness by measuring the loss in property dispersion as a dataset is progressively coarse-grained through hierarchical clustering. However, ROGI values are not comparable across representations of different dimensionalities because distances between randomly sampled points increase with dimension, artificially deflating ROGI for high-dimensional representations.
ROGI-XD addresses this by changing the integration variable. Instead of integrating over normalized distance threshold $t$, ROGI-XD integrates over $1 - \log N_{\text{clusters}} / \log N$, where $N_{\text{clusters}}$ is the number of clusters at a given dendrogram step and $N$ is the dataset size. This variable captures the degree of coarse-graining independent of representation dimensionality, producing comparable roughness values across representations ranging from 14 dimensions (descriptors) to 2048 dimensions (ChemGPT).
The procedure follows five steps: (1) cluster molecules using complete linkage at distance threshold $t$, (2) coarse-grain by replacing each property label $y_i$ with its cluster mean $\bar{y}_j$, (3) compute the standard deviation $\sigma_t$ of the coarse-grained dataset, (4) repeat for all dendrogram steps, and (5) compute the area under the curve of $2(\sigma_0 - \sigma_t)$ versus the new integration variable.
Representations and Tasks Evaluated
The study compares seven molecular representations:
| Representation | Type | Dimensionality | Source |
|---|---|---|---|
| Descriptors | Fixed | 14 | RDKit (14 properties) |
| Morgan FP | Fixed | 512 | Radius 2, 512-bit |
| VAE | Pretrained | 128 | Character-based SMILES VAE, ZINC 250k |
| GIN | Pretrained | 300 | Node attribute masking, ZINC 250k |
| ChemBERTa | Pretrained | 384 | 77M molecules, masked LM |
| ChemGPT | Pretrained | 2048 | PubChem 10M, causal LM |
| Random | Baseline | 128 | Uniform $[0,1]^{128}$ |
These are evaluated on 17 regression tasks drawn from two sources: ADMET datasets from the Therapeutics Data Commons (TDC) and toy datasets generated using GuacaMol oracle functions. Five ML models are used for cross-validation: KNN, MLP, PLS, random forest, and SVR.
Pretrained Representations Are Not Smoother
ROGI-XD correlates strongly with cross-validated RMSE across representations (median Pearson $r = 0.72$-$0.88$ depending on model), compared to the original ROGI which produces weak cross-representation correlations (median $r \in [-0.32, 0.28]$). When correlating over both representations and tasks simultaneously, ROGI-XD achieves $r = 0.91$-$0.99$ versus $r = 0.68$-$0.84$ for the original ROGI.
Using this validated metric, the authors find that pretrained representations do not produce smoother QSPR surfaces than fingerprints or descriptors. In more than 50% of tasks, both descriptors and fingerprints generate smoother surfaces. The median relative ROGI-XD increase for pretrained representations is 9.1-21.3% compared to descriptors and 2.3-10.1% compared to fingerprints, indicating rougher surfaces.
As a practical tool, ROGI-XD can guide representation selection without exhaustive benchmarking. Selecting the representation with the lowest ROGI-XD for each task and then optimizing over model architecture results in only a 6.8% average relative increase in best-case model error across the 17 tasks. In 8 of 17 tasks, the lowest ROGI-XD correctly identifies the optimal representation.
Fine-tuning can improve smoothness. On the Lipophilicity task ($N_{\text{tot}} = 4200$), fine-tuning the VAE with a contrastive loss reduces ROGI-XD from 0.254 to 0.107 ($\pm 0.02$), well below the descriptor baseline of 0.227. On the smaller CACO2 task ($N_{\text{tot}} = 910$), fine-tuning yields ROGI-XD of 0.143 ($\pm 0.05$), comparable to descriptors at 0.132. The impact of fine-tuning is sensitive to both the task and the amount of labeled data.
Implications for Chemical Foundation Model Development
The lack of smoothness in pretrained QSPR surfaces explains the inconsistent empirical performance of chemical foundation models. The authors note that ROGI-XD is thematically similar to a contrastive loss, as both scale proportionally with the frequency and severity of activity cliffs. This connection suggests that imposing stronger smoothness assumptions during pretraining, for example through weak supervision on calculable molecular properties, could help produce representations that generalize better to downstream property prediction. ROGI-XD provides a practical tool for evaluating new pretraining strategies without exhaustive benchmark testing: a representation with lower ROGI-XD on a given task is likely to yield lower model error.
A limitation is that the study treats pretrained representations as static (frozen features). Fine-tuning introduces many additional design choices and can substantially improve representation quality, but this evaluation is left for future work. Additionally, the survey of pretrained models is not exhaustive and focuses on four representative architectures.
Reproducibility
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| coleygroup/rogi-xd | Code | MIT | Official implementation with pretrained models and notebooks; results reproducible via make all |
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining (VAE, GIN) | ZINC 250k | 250,000 | 80/20 train/val split |
| Pretraining (ChemBERTa) | PubChem | 77M | Masked language modeling |
| Pretraining (ChemGPT) | PubChem 10M | 10M | Causal language modeling |
| Evaluation | TDC ADMET | ~900-10,000 per task | 12 regression tasks |
| Evaluation | GuacaMol oracles | 10,000 per task | 5 synthetic tasks |
Algorithms
- ROGI-XD: Hierarchical clustering (complete linkage) with integration over $1 - \log N_{\text{clusters}} / \log N$
- Cross-validation: 5-fold CV with KNN, MLP, PLS, RF (n_estimators=50), SVR from scikit-learn
- Fine-tuning loss: $\mathscr{L} = \mathscr{L}_{\text{CE}} + \beta \cdot \mathscr{L}_{\text{KL}} + \gamma \cdot \mathscr{L}_{\text{cont}}$ with $\beta = 0.1$, $\gamma = 50$; contrastive term uses cosine distance in latent space and absolute value in target space
Hardware
Two AMD Ryzen Threadripper PRO 3995WX CPUs, four NVIDIA A5000 GPUs, 512 GB RAM, Ubuntu 20.04 LTS.
Paper Information
Citation: Graff, D. E., Pyzer-Knapp, E. O., Jordan, K. E., Shakhnovich, E. I., & Coley, C. W. (2023). Evaluating the roughness of structure-property relationships using pretrained molecular representations. Digital Discovery, 2(5), 1452-1460. https://doi.org/10.1039/d3dd00088e
Publication: Digital Discovery 2023
Additional Resources:
Citation
@article{graff2023roughness,
title={Evaluating the roughness of structure--property relationships using pretrained molecular representations},
author={Graff, David E. and Pyzer-Knapp, Edward O. and Jordan, Kirk E. and Shakhnovich, Eugene I. and Coley, Connor W.},
journal={Digital Discovery},
volume={2},
number={5},
pages={1452--1460},
year={2023},
publisher={Royal Society of Chemistry},
doi={10.1039/d3dd00088e}
}