A Unified Resource for Generative Molecular Design
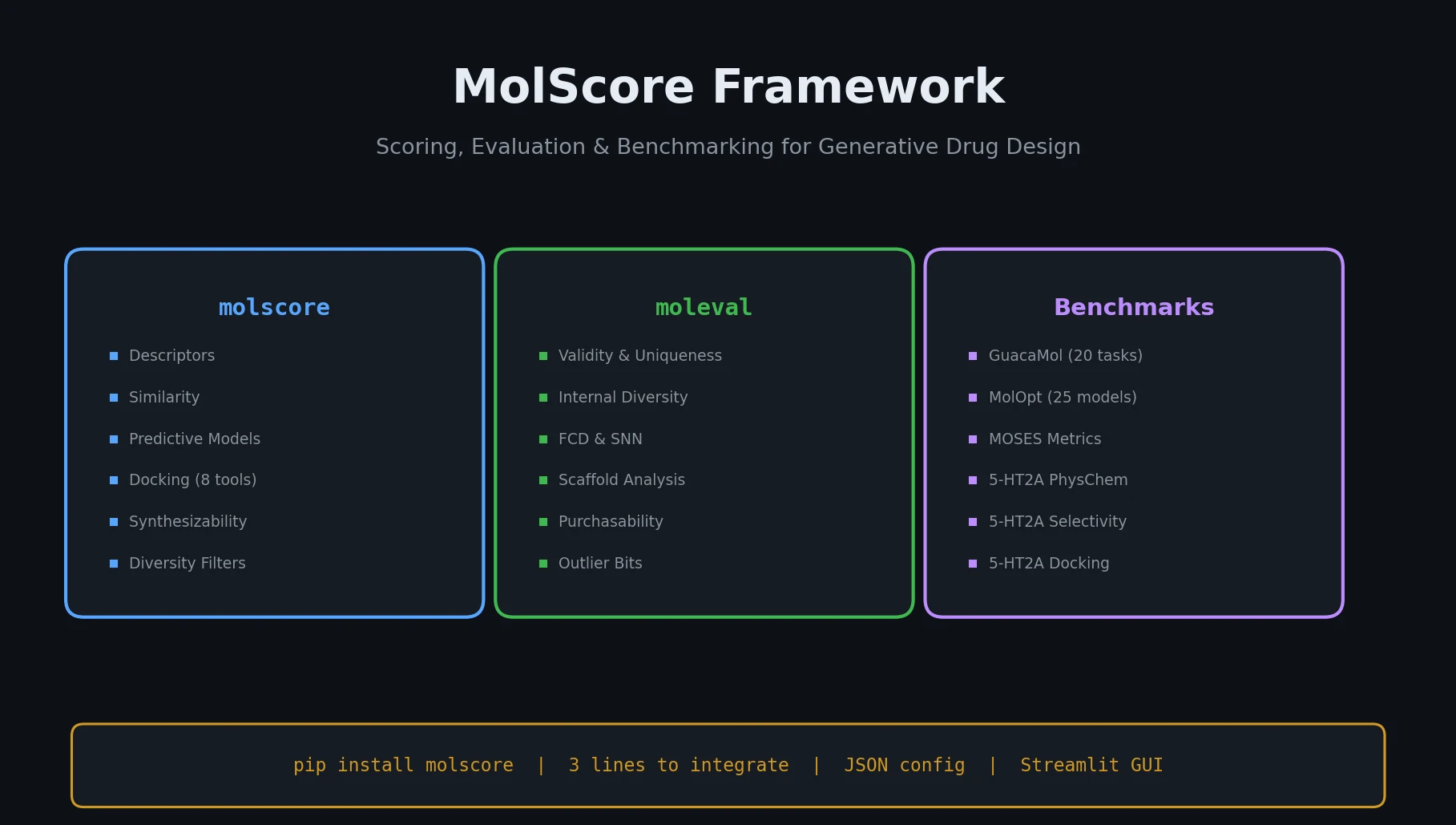
MolScore is a Resource paper that introduces an open-source Python framework for scoring, evaluating, and benchmarking generative models in de novo drug design. The primary contribution is the software itself: a modular, configurable platform that consolidates functionality previously scattered across multiple tools (GuacaMol, MOSES, MolOpt, REINVENT, TDC) into a single package. MolScore provides scoring functions for molecular optimization, evaluation metrics for assessing the quality of generated molecules, and a benchmark mode for standardized comparison of generative models.
The Fragmented Landscape of Generative Model Evaluation
Generative models for molecular design have proliferated rapidly, but evaluating and comparing them remains difficult. Existing benchmarks each address only part of the problem:
- GuacaMol provides 20 fixed optimization objectives but cannot separate top-performing models on most tasks, and custom objectives require code modification.
- MOSES focuses on distribution-learning metrics but does not support molecular optimization.
- MolOpt extends benchmark evaluation to 25 generative approaches but lacks evaluation of the quality of generated chemistry.
- Docking benchmarks (smina-docking-benchmark, DOCKSTRING, TDC) test structure-based scoring but often lack proper ligand preparation, leading generative models to exploit non-holistic objectives by generating large or greasy molecules.
- REINVENT provides configurable scoring functions but is tightly coupled to its own generative model architecture.
No single tool offered configurable objectives, comprehensive evaluation metrics, generative-model-agnostic design, and graphical user interfaces together. This fragmentation forces practitioners to write custom glue code and makes reproducible comparison across methods difficult.
Modular Architecture for Scoring, Evaluation, and Benchmarking
MolScore is split into two sub-packages:
molscore: Molecule Scoring
The molscore sub-package handles iterative scoring of SMILES generated by any generative model. The workflow for each iteration:
- Parse and validate SMILES via RDKit, canonicalize, and check intra-batch uniqueness.
- Cross-reference against previously generated molecules to reuse cached scores (saving compute for expensive scoring functions like docking).
- Run user-specified scoring functions on valid, unique molecules (invalid molecules receive a score of 0).
- Transform each score to a 0-1 range using configurable transformation functions (normalize, linear threshold, Gaussian threshold, step threshold).
- Aggregate transformed scores into a single desirability score using configurable aggregation (weighted sum, product, geometric mean, arithmetic mean, Pareto front, or auto-weighted variants).
- Optionally apply diversity filters to penalize non-diverse molecules, or use any scoring function as a multiplicative filter.
The full objective is specified in a single JSON configuration file, with a Streamlit GUI provided for interactive configuration writing. The available scoring functions span:
| Category | Examples |
|---|---|
| Descriptors | RDKit descriptors, linker descriptors, penalized logP |
| Similarity | Fingerprint similarity, ROCS, Open3DAlign, substructure matching |
| Predictive models | Scikit-learn models, PIDGINv5 (2,337 ChEMBL31 targets), ChemProp, ADMET-AI |
| Docking | Glide, PLANTS, GOLD, OEDock, Smina, Gnina, Vina, rDock |
| Synthesizability | SA score, RA Score, AiZynthFinder, reaction filters |
Most scoring functions support multiprocessing, and computationally expensive functions (docking, ligand preparation) can be distributed across compute clusters via Dask.
moleval: Molecule Evaluation
The moleval sub-package computes performance metrics on generated molecules relative to reference datasets. It extends the MOSES metric suite with additional intrinsic metrics (sphere exclusion diversity, scaffold uniqueness, functional group and ring system diversity, ZINC20 purchasability via molbloom) and extrinsic metrics (analogue similarity/coverage, functional group and ring system similarity, outlier bits or “Silliness”).
Benchmark Mode
A MolScoreBenchmark class iterates over a list of JSON configuration files, providing standardized comparison. Pre-built presets reimplement GuacaMol and MolOpt benchmarks, and users can define custom benchmark suites without writing code.
Case Studies: 5-HT2A Ligand Design and Fine-Tuning Evaluation
The authors demonstrate MolScore with a SMILES-based RNN generative model using Augmented Hill-Climb for optimization, designing serotonin 5-HT2A receptor ligands across three objective sets of increasing complexity.
First Objective Set: Basic Drug Properties
Four objectives combine predicted 5-HT2A activity (via PIDGINv5 random forest models at 1 uM) with synthesizability (RAscore) and/or BBB permeability property ranges (TPSA < 70, HBD < 2, logP 2-4, MW < 400). All objectives were optimized successfully, with diversity filters preventing mode collapse. The most difficult single objective (5-HT2A activity alone) was hardest primarily because the diversity filter more heavily penalized similar molecules for this relatively easy task.
Second Objective Set: Selectivity
Six objectives incorporate selectivity proxies using PIDGINv5 models for off-target prediction against Class A GPCR membrane receptors (266 models), the D2 dopamine receptor, dopamine receptor family, serotonin receptor subtypes, and combinations. These proved substantially harder: selectivity against dopamine and serotonin receptor families combined was barely improved during optimization. Even with imperfect predictive models, the PIDGINv5 ensemble correctly identified 95 of 126 known selective 5-HT2A ligands. Nearest-neighbor analysis of de novo molecules (Tanimoto similarity 0.3-0.6) showed they tended to be structurally simpler versions of known selective ligands.
Third Objective Set: Structure-Based Docking
Two objectives use molecular docking via GlideSP into 5-HT2A (PDB: 6A93) and D2 (PDB: 6CM4) crystal structures with full ligand preparation (LigPrep for stereoisomer/tautomer/protonation state enumeration). Multi-parameter optimization includes docking score, D155 polar interaction constraint, formal charge, and consecutive rotatable bond limits. Single-target docking scores reached the mean of known ligands within 200 steps, but optimizing for divergent 5-HT2A vs D2 docking scores was much harder due to binding pocket similarity. Protein-ligand interaction fingerprint analysis (ProLIF) revealed that molecules optimized for selectivity avoided specific binding pocket regions shared between the two receptors.
Evaluation Case Study: Fine-Tuning Epochs
The moleval sub-package was used to track metrics across fine-tuning epochs of a SMILES RNN on A2A receptor ligands, showing that just one or two epochs sufficed to increase similarity to the fine-tuning set, while further epochs reduced novelty and diversity.
Configurable Benchmarking with Practical Drug Design Relevance
MolScore provides a more comprehensive platform than any single existing tool. Compared to prior work:
| Feature | GuacaMol | MOSES | MolOpt | TDC | REINVENT | MolScore |
|---|---|---|---|---|---|---|
| Configurable objectives | No | N/A | No | No | Yes | Yes |
| Optimization objectives | Yes | No | Yes | Yes | Yes | Yes |
| Evaluation metrics | Yes | Yes | No | No | No | Yes |
| Model-agnostic | Yes | Yes | Yes | Yes | No | Yes |
| GUI | No | No | No | No | Yes | Yes |
The framework integrates into any Python-based generative model in three lines of code. Dependency conflicts between scoring function libraries are handled by running conflicting components as local servers from isolated conda environments.
Key limitations acknowledged by the authors include: the assumption of conda for environment management, the inherent difficulty of designing non-exploitable objectives, and the fact that ligand-based predictive models may have limited applicability domains for out-of-distribution de novo molecules.
Future directions include accepting 3D molecular conformations as inputs, structure interaction fingerprint rescoring, and dynamic configuration files for curriculum learning.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pre-training | ChEMBL compounds | Not specified | Standard ChEMBL training set for SMILES RNN |
| Evaluation reference | 5-HT2A ligands from ChEMBL31 | 3,771 compounds | Extracted for score distribution comparison |
| Activity models | PIDGINv5 on ChEMBL31 | 2,337 target models | Random forest classifiers at various concentration thresholds |
| Fine-tuning | A2A receptor ligands | Not specified | Used for moleval case study |
Algorithms
The generative model used in case studies is a SMILES-based RNN with Augmented Hill-Climb reinforcement learning. Diversity filters penalize non-diverse molecules during optimization. Score transformation functions (normalize, linear threshold, Gaussian threshold, step threshold) map raw scores to 0-1 range. Aggregation functions (arithmetic mean, weighted sum, product, geometric mean, Pareto front) combine multi-parameter objectives.
Models
PIDGINv5 provides 2,337 pre-trained random forest classifiers on ChEMBL31 targets. RAscore provides pre-trained synthesizability prediction. ADMET-AI and ChemProp models are supported via isolated environments. Docking uses GlideSP with LigPrep for ligand preparation in the structure-based case study.
Evaluation
Intrinsic metrics: validity, uniqueness, scaffold uniqueness, internal diversity, sphere exclusion diversity, Solow-Polasky diversity, scaffold diversity, functional group diversity, ring system diversity, MCF and PAINS filters, ZINC20 purchasability.
Extrinsic metrics: novelty, FCD, analogue similarity/coverage, functional group similarity, ring system similarity, SNN similarity, fragment similarity, scaffold similarity, outlier bits, Wasserstein distance on LogP/SA/NP/QED/MW.
Hardware
Not specified in the paper. Docking-based objectives can be distributed across compute clusters via Dask.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| MolScore | Code | MIT | Main framework, installable via pip |
| MolScore Examples | Code | MIT | Integration examples with SMILES-RNN, CReM, GraphGA |
Paper Information
Citation: Thomas, M., O’Boyle, N. M., Bender, A., & de Graaf, C. (2024). MolScore: a scoring, evaluation and benchmarking framework for generative models in de novo drug design. Journal of Cheminformatics, 16(1), 64. https://doi.org/10.1186/s13321-024-00861-w
@article{thomas2024molscore,
title={MolScore: a scoring, evaluation and benchmarking framework for generative models in de novo drug design},
author={Thomas, Morgan and O'Boyle, Noel M. and Bender, Andreas and de Graaf, Chris},
journal={Journal of Cheminformatics},
volume={16},
number={1},
pages={64},
year={2024},
publisher={BioMed Central},
doi={10.1186/s13321-024-00861-w}
}