A Comprehensive Benchmark for Structure-Based Molecular Generation
MolGenBench is a Resource paper that provides a large-scale, application-oriented benchmark for evaluating molecular generative models in the context of structure-based drug design (SBDD). The primary contribution is a dataset of 220,005 experimentally validated active molecules across 120 protein targets, organized into 5,433 chemical series, along with a suite of novel evaluation metrics. The benchmark addresses both de novo molecular design and hit-to-lead (H2L) optimization, a critical drug discovery stage that existing benchmarks largely ignore.
Gaps in Existing Molecular Generation Benchmarks
Despite rapid progress in deep generative models for drug discovery, the evaluation landscape has not kept pace. The authors identify four categories of limitations in existing benchmarks:
Dataset construction: Existing benchmarks use overly stringent activity cutoffs and too few protein targets. The widely used CrossDocked2020 dataset contains very few reference ligands per target, making it difficult to evaluate whether a model can rediscover the full distribution of active compounds.
Model selection: Prior benchmark studies evaluate a narrow range of architectures and do not systematically examine the effects of training data composition, prior knowledge integration, or architectural paradigm.
Evaluation scenarios: Existing benchmarks focus exclusively on de novo generation. Hit-to-lead optimization, where a hit compound is refined through R-group modifications, remains unstandardized.
Evaluation metrics: Standard metrics (QED, Vina score, SA score) correlate strongly with atom count and fail to assess target-specific generation capacity. The AddCarbon model illustrates this: simply adding random carbon atoms to training molecules achieves near-perfect scores on standard metrics while producing nonsensical chemistry.
Novel Metrics for Evaluating Molecular Generation
MolGenBench introduces three key metrics designed to capture aspects of model performance that existing metrics miss.
Target-Aware Score (TAScore)
The TAScore measures whether a model generates target-specific molecules rather than generic structures. It compares the ratio of active molecule or scaffold recovery on a specific target to the background recovery across all targets:
$$ \text{TAScore}_{\text{label}, i} = \frac{S_{i} / S_{\text{all}}}{R_{i} / R_{\text{all}}}; \quad \text{label} \in \{\text{SMILES}, \text{scaffold}\} $$
For target $i$: $R_{\text{all}}$ is the total number of distinct molecules generated across all 120 targets; $R_{i}$ is the subset matching known actives for target $i$ (without conditioning on target $i$); $S_{\text{all}}$ is the total generated when conditioned on target $i$; and $S_{i}$ is the subset matching known actives for target $i$. A TAScore above 1 indicates the model uses target-specific information effectively.
Hit Rate
The hit rate quantifies the efficiency of active compound discovery:
$$ \text{HitRate}_{\text{label}} = \frac{\mathcal{M}_{\text{active}}}{\mathcal{M}_{\text{sampled}}}; \quad \text{label} \in \{\text{SMILES}, \text{scaffold}\} $$
where $\mathcal{M}_{\text{active}}$ is the number of unique active molecules or scaffolds found, and $\mathcal{M}_{\text{sampled}}$ is the total number of generated molecules.
Mean Normalized Affinity (MNA) Score
For H2L optimization, the MNA Score measures whether models generate compounds with improved potency relative to the known activity range within each chemical series:
$$ \text{NA}_{g} = \frac{\text{Affinity}_{g}^{\text{series}} - \text{Affinity}_{\min}^{\text{series}}}{\text{Affinity}_{\max}^{\text{series}} - \text{Affinity}_{\min}^{\text{series}}} $$
$$ \text{MNAScore} = \frac{1}{G} \sum_{g}^{G} \text{NA}_{g} $$
This normalizes affinities to [0, 1] within each series, enabling cross-series comparison.
Systematic Evaluation of 17 Generative Models Across Two Drug Discovery Scenarios
Dataset Construction
The MolGenBench dataset was built from ChEMBL v33. Ligands failing RDKit validation were discarded, along with entries where binding affinity exceeded 10 uM. The 120 protein targets were selected based on minimum thresholds: at least 50 active molecules, at least 50 unique Bemis-Murcko scaffolds, and at least 20 distinct chemical series per target. For H2L optimization, maximum common substructures (MCS) were identified per series, with dual thresholds requiring the MCS to appear in over 80% of molecules and cover more than one-third of each molecule’s atoms. The top 5 series per target (ranked by dockable ligands) formed the H2L test set: 600 compound series across 120 targets.
Evaluated Models
De novo models (10): Pocket2Mol, TargetDiff, FLAG, DecompDiff, SurfGen, PocketFlow, MolCraft, TamGen, DiffSBDD-M (trained on BindingMOAD), DiffSBDD-C (trained on CrossDock). These span autoregressive, diffusion, and Bayesian flow network architectures.
H2L models (7): Fragment-based (DiffSBDD-M/C inpainting, Delete, DiffDec) and ligand-based (ShEPhERD, ShapeMol, PGMG). These use pharmacophore, surface, or shape priors.
Models were further stratified by whether test proteins appeared in their CrossDock training set (“Proteins in CrossDock” vs. “Proteins Not in CrossDock”), enabling direct measurement of generalization.
Evaluation Dimensions
The benchmark evaluates six dimensions:
| Dimension | Key Metrics |
|---|---|
| Basic molecular properties | Validity, QED, SA score, uniqueness, diversity, JSD alignment |
| Chemical safety | Industry-standard filter pass rates (Eli Lilly, Novartis, ChEMBL rules) |
| Conformational quality | PoseBusters pass rate, strain energy, steric clash frequency |
| Active compound recovery | Hit rate, hit fraction, active molecule and scaffold recovery counts |
| Target awareness | TAScore at molecule and scaffold levels |
| Lead optimization | MNA Score, number of series with hits |
Key Results: Basic Properties and Chemical Safety
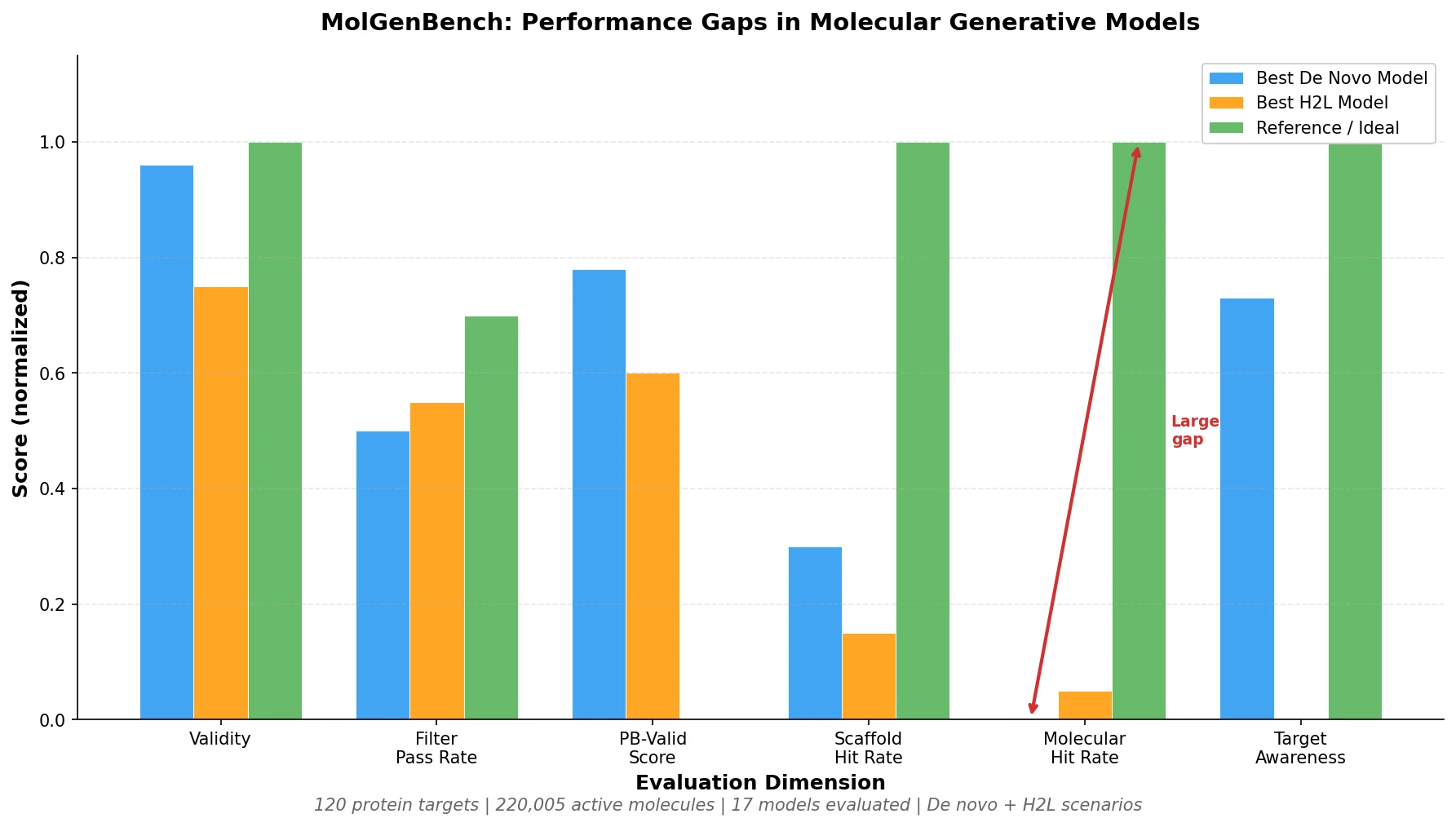
Most models generate drug-like molecules with reasonable QED (0.4-0.6) and SA scores (0.5-0.8). However, two models (FLAG, SurfGen) showed validity below 0.4. TamGen exhibited low uniqueness (~27%), suggesting overreliance on pretrained patterns.
Chemical filter pass rates revealed a more concerning picture: only TamGen and PGMG exceeded 50% of molecules passing all industry-standard filters. Most models fell below 40%, and some (FLAG, SurfGen) below 5%. Nearly 70% of reference active molecules passed the same filters, indicating models frequently generate high-risk compounds.
Key Results: Conformational Quality
MolCraft achieved the highest PoseBusters validity (0.783 PB-valid score among valid molecules). PocketFlow, despite perfect SMILES validity, had fewer than half of its valid molecules pass conformational checks. Most models produced conformations with higher strain energy than those from AutoDock Vina. Some models (MolCraft for de novo, DiffDec for H2L) surpassed Vina in minimizing steric clashes, suggesting advanced architectures can exceed the patterns in their training data.
Key Results: Active Compound Recovery and Hit Rates
De novo models exhibited very low hit rates. The highest molecular hit rate among de novo models was 0.124% on proteins in CrossDock, dropping to 0.024% on unseen proteins. Scaffold-level hit rates were 10-fold higher, showing that generating pharmacologically plausible scaffolds is considerably easier than generating fully active molecules.
After removing molecules overlapping with the CrossDock training set, TamGen’s recovery dropped substantially (from 30.3 to 18.7 targets), confirming significant memorization effects. On proteins not in CrossDock, half of the de novo models failed to recover any active molecules at all.
Fragment-based H2L models substantially outperformed both de novo models and ligand-based H2L approaches. Delete recovered active molecules in 44.3 series (out of 600), and DiffDec in 34.7 series.
Key Results: Target Awareness
Most de novo models failed the TAScore evaluation. PocketFlow showed the strongest target awareness at the scaffold level, with only 27% of targets showing TAScore < 1 (indicating no target specificity). At the molecular level, results were even weaker: TamGen achieved TAScore > 1 for only 30.6% of CrossDock-seen targets and just 4 out of 35 unseen targets. Most models generated structurally similar molecules regardless of which target they were conditioned on.
Key Results: H2L Optimization (MNA Score)
DiffDec achieved the highest total active hits (121.7) and the best MNA Score (0.523), followed by Delete (104.7 hits, MNA Score 0.482). Ligand-based models (ShEPhERD, PGMG) recovered fewer hits but showed higher MNA Scores per hit, suggesting pharmacophore-based priors help prioritize more potent molecules when actives are found. The most successful model (Delete) achieved a hit in only 9.6% of series (57/600), indicating substantial room for improvement.
Critical Findings and Limitations of Current Molecular Generative Models
The benchmark reveals several consistent limitations:
Low screening efficiency: De novo models achieve molecular hit rates below 0.13%, far from practical utility. Scaffold recovery is more feasible but still limited.
Weak target awareness: Most SBDD models fail to use protein structural information effectively, generating similar molecules across different targets. This raises concerns about off-target effects.
Conformational prediction remains difficult: Most models produce conformations with higher strain energy than classical docking, and only a small fraction (typically below 23%) of generated poses match redocked conformations within 2 Angstrom RMSD.
Generalization gap: Performance consistently drops on proteins not in the training set, and prior benchmarks that do not stratify by training data exposure overestimate real-world utility.
Inference-time scaling does not solve the problem: Sampling up to 100,000 molecules increased the absolute number of active discoveries but with diminishing efficiency. Without better scoring functions, scaling sampling offers limited practical value.
Chemical safety: Most models produce a majority of molecules that fail industry-standard reactivity and promiscuity filters.
The authors acknowledge that the benchmark’s 220,005 active molecules represent a biased subset of bioactive chemical space. A model’s failure to rediscover known actives for a given target may reflect sampling limitations rather than generating inactive compounds.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Active compounds | ChEMBL v33 | 220,005 molecules, 120 targets | Filtered at 10 uM affinity threshold |
| H2L series | ChEMBL v33 + PDB | 5,433 series (600 used for H2L test) | MCS-based series construction |
| Protein structures | PDB | 120 targets | One PDB entry per target |
| Training (most models) | CrossDocked2020 | Varies | Standard SBDD training set |
Algorithms
- De novo models sampled 1,000 molecules per target; H2L models sampled 200 per series
- All experiments repeated three times with different random seeds
- Docking performed with AutoDock Vina using standard parameters
- Chemical filters applied via the medchem library
- Conformational quality assessed with PoseBusters and PoseCheck
- Interaction scores computed via ProLIF with frequency-weighted normalization
Models
All 17 models were obtained from their official GitHub repositories and run with default configurations. The benchmark does not introduce new model architectures.
Evaluation
Summary of key metrics across the best-performing models in each category:
| Metric | Best De Novo | Value | Best H2L | Value |
|---|---|---|---|---|
| PB-valid score | MolCraft | 0.783 | DiffSBDD-M | 0.597 |
| Molecular hit rate (in CrossDock) | TamGen | 0.124% | DiffDec | Higher than de novo |
| Scaffold hit rate (in CrossDock) | PocketFlow | >10% | Delete | Lower than PocketFlow |
| TAScore scaffold (% targets >1) | PocketFlow | 73% | N/A | N/A |
| MNA Score | N/A | N/A | DiffDec | 0.523 |
| Filter pass rate | TamGen | >50% | PGMG | >50% |
Hardware
Specific hardware requirements are not detailed in the paper. Models were run using their default configurations from official repositories.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| MolGenBench | Code | MIT | Benchmark evaluation framework |
| Zenodo dataset | Dataset | CC-BY-NC-ND 4.0 | Processed data and source data for all results |
Paper Information
Citation: Cao, D., Fan, Z., Yu, J., Chen, M., Jiang, X., Sheng, X., Wang, X., Zeng, C., Luo, X., Teng, D., & Zheng, M. (2025). Benchmarking Real-World Applicability of Molecular Generative Models from De novo Design to Lead Optimization with MolGenBench. bioRxiv. https://doi.org/10.1101/2025.11.03.686215
@article{cao2025molgenbench,
title={Benchmarking Real-World Applicability of Molecular Generative Models from De novo Design to Lead Optimization with MolGenBench},
author={Cao, Duanhua and Fan, Zhehuan and Yu, Jie and Chen, Mingan and Jiang, Xinyu and Sheng, Xia and Wang, Xingyou and Zeng, Chuanlong and Luo, Xiaomin and Teng, Dan and Zheng, Mingyue},
journal={bioRxiv},
year={2025},
doi={10.1101/2025.11.03.686215}
}