A Resource Paper for Molecular Machine Learning Benchmarking
This is a Resource paper. MoleculeNet provides a standardized benchmark suite for evaluating molecular machine learning methods. Its primary contribution is the curation of 17 public datasets spanning four categories of molecular properties, together with standardized evaluation metrics, multiple dataset splitting strategies, and open-source implementations of featurization and learning algorithms via the DeepChem library.
Why Molecular ML Needed a Unified Benchmark
Prior to MoleculeNet, algorithmic progress in molecular machine learning was difficult to measure. Individual papers benchmarked proposed methods on different datasets with different metrics, making cross-method comparison unreliable. Several factors make molecular ML particularly challenging:
- Data scarcity: Molecular datasets are much smaller than those available for computer vision or NLP, since obtaining accurate chemical property measurements requires specialized instruments and expert supervision.
- Heterogeneous outputs: Properties of interest range from quantum mechanical characteristics to macroscopic physiological effects on the human body.
- Variable input structures: Molecules have arbitrary size, variable connectivity, and many possible 3D conformers, all of which must be encoded into fixed-length representations for conventional ML algorithms.
- No standard evaluation protocol: Without prescribed metrics, splits, or data subsets, two methods using the same underlying database (e.g., PubChem) could be entirely incomparable.
Existing databases like PubChem, ChEMBL, and the Quantum Machine collections provided raw data but did not define evaluation protocols suitable for machine learning development. MoleculeNet bridges this gap, following the precedent set by ImageNet in computer vision and WordNet in NLP.
Core Design: Datasets, Splits, Metrics, and Featurizations
MoleculeNet is organized around four components: curated datasets, splitting methods, evaluation metrics, and molecular featurizations.
Datasets Across Four Property Categories
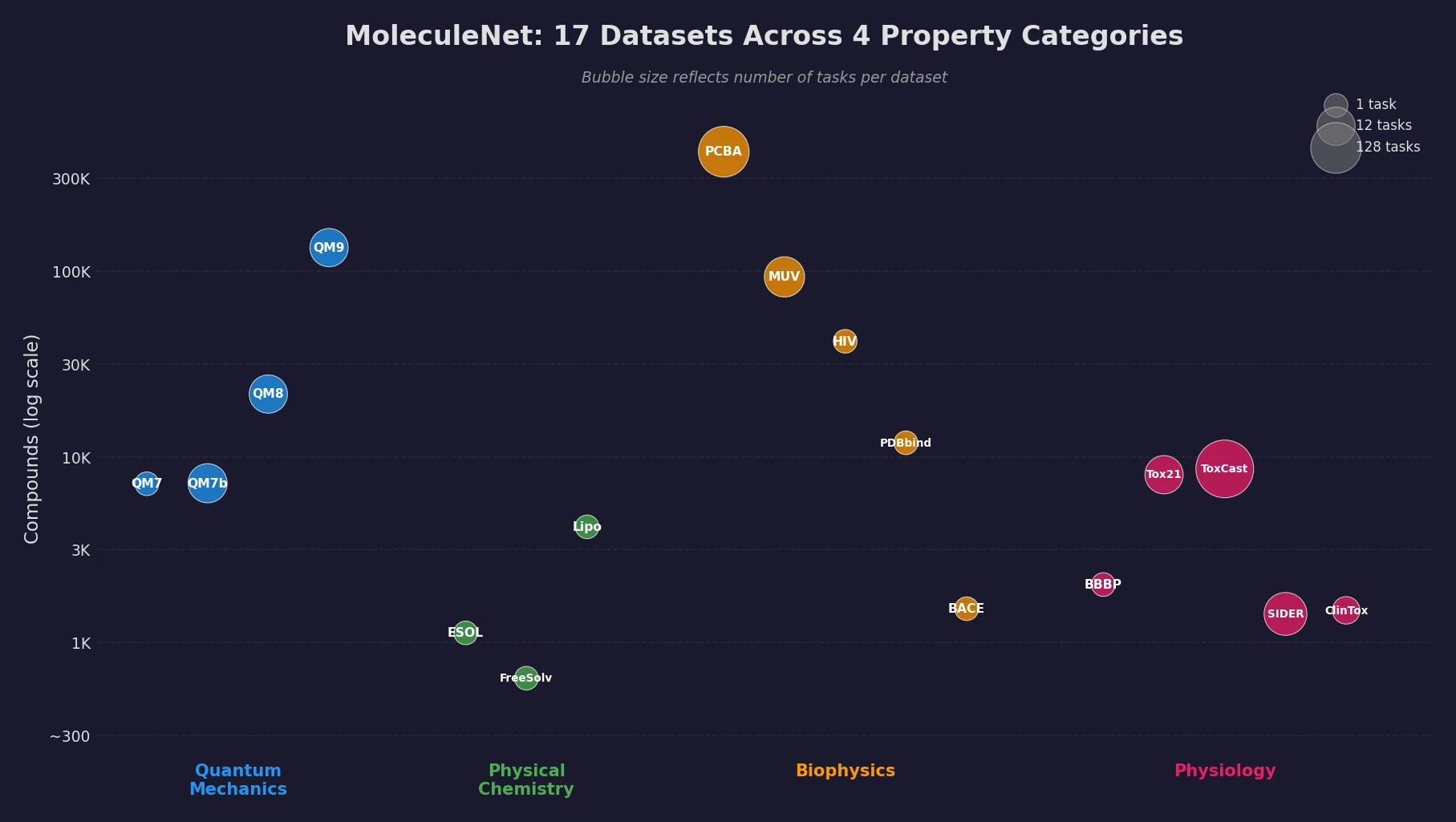
The benchmark includes 17 datasets covering over 700,000 compounds and more than 800 tasks. These are organized into four categories reflecting different levels of molecular properties:
| Category | Dataset | Tasks | Compounds | Task Type | Rec. Split | Rec. Metric |
|---|---|---|---|---|---|---|
| Quantum Mechanics | QM7 | 1 | 7,165 | Regression | Stratified | MAE |
| QM7b | 14 | 7,211 | Regression | Random | MAE | |
| QM8 | 12 | 21,786 | Regression | Random | MAE | |
| QM9 | 12 | 133,885 | Regression | Random | MAE | |
| Physical Chemistry | ESOL | 1 | 1,128 | Regression | Random | RMSE |
| FreeSolv | 1 | 643 | Regression | Random | RMSE | |
| Lipophilicity | 1 | 4,200 | Regression | Random | RMSE | |
| Biophysics | PCBA | 128 | 439,863 | Classification | Random | PRC-AUC |
| MUV | 17 | 93,127 | Classification | Random | PRC-AUC | |
| HIV | 1 | 41,913 | Classification | Scaffold | ROC-AUC | |
| PDBbind | 1 | 11,908 | Regression | Time | RMSE | |
| BACE | 1 | 1,522 | Classification | Scaffold | ROC-AUC | |
| Physiology | BBBP | 1 | 2,053 | Classification | Scaffold | ROC-AUC |
| Tox21 | 12 | 8,014 | Classification | Random | ROC-AUC | |
| ToxCast | 617 | 8,615 | Classification | Random | ROC-AUC | |
| SIDER | 27 | 1,427 | Classification | Random | ROC-AUC | |
| ClinTox | 2 | 1,491 | Classification | Random | ROC-AUC |
Quantum mechanics datasets (QM7, QM7b, QM8, QM9) contain DFT-computed electronic properties for subsets of the GDB database. Physical chemistry datasets cover solubility (ESOL), hydration free energy (FreeSolv), and lipophilicity. Biophysics datasets include high-throughput screening results (PCBA, MUV), HIV inhibition activity, protein-ligand binding affinity (PDBbind), and BACE-1 inhibition. Physiology datasets cover blood-brain barrier penetration (BBBP), toxicity (Tox21, ToxCast), side effects (SIDER), and clinical trial toxicity (ClinTox).
Data Splitting Strategies
MoleculeNet implements four splitting methods, all using an 80/10/10 train/validation/test ratio:
- Random splitting: Standard random assignment to subsets.
- Scaffold splitting: Separates molecules by their 2D structural frameworks (Bemis-Murcko scaffolds), providing a harder generalization test since structurally different molecules appear in different subsets.
- Stratified splitting: Ensures each subset contains the full range of label values (used for QM7).
- Time splitting: Trains on older data and tests on newer data to mimic real-world development (used for PDBbind).
Evaluation Metrics
Regression tasks use MAE or RMSE depending on the dataset. Classification tasks use either ROC-AUC or PRC-AUC. The choice between ROC-AUC and PRC-AUC depends on class imbalance: PRC-AUC is recommended for datasets with positive rates below 2% (PCBA, MUV), since precision-recall curves better capture performance under extreme imbalance.
The false positive rate and precision are defined as:
$$ \text{FPR} = \frac{\text{false positive}}{\text{false positive} + \text{true negative}} $$
$$ \text{precision} = \frac{\text{true positive}}{\text{false positive} + \text{true positive}} $$
When positive samples form a small fraction of the data, false positives influence precision much more than FPR, making PRC-AUC more informative than ROC-AUC.
Featurization Methods
MoleculeNet implements six molecular featurization approaches:
- ECFP (Extended-Connectivity Fingerprints): Fixed-length binary fingerprints capturing topological substructures via hashing.
- Coulomb Matrix: Encodes nuclear charges and 3D coordinates through atomic self-energies and Coulomb repulsion:
$$ M_{IJ} = \begin{cases} 0.5 Z_{I}^{2.4} & \text{for } I = J \\ \frac{Z_{I} Z_{J}}{|\mathbf{R}_{I} - \mathbf{R}_{J}|} & \text{for } I \neq J \end{cases} $$
- Grid Featurizer: Designed for PDBbind, incorporating both ligand and protein structural information including salt bridges, hydrogen bonds, and SPLIF fingerprints.
- Symmetry Functions: Preserve rotational and permutation symmetry through radial and angular functions between atom pairs and triplets.
- Graph Convolutions: Compute initial atom feature vectors and neighbor lists from molecular graphs.
- Weave: Similar to graph convolutions but also computes pairwise atom features encoding bond properties, graph distance, and ring information.
Benchmarked Models and Experimental Setup
MoleculeNet benchmarks 12 learning algorithms divided into conventional methods and graph-based methods.
Conventional Methods
- Logistic Regression (classification only)
- Kernel SVM with radial basis function kernel
- Kernel Ridge Regression (KRR)
- Random Forests
- Gradient Boosting (XGBoost)
- Singletask/Multitask Networks: Fully connected networks with shared layers across tasks
- Bypass Networks: Multitask networks augmented with per-task “bypass” layers that directly connect inputs to outputs
- Influence Relevance Voting (IRV): Refined K-nearest neighbor classifiers using Jaccard-Tanimoto similarity:
$$ S(\vec{A}, \vec{B}) = \frac{A \cap B}{A \cup B} $$
Graph-Based Methods
- Graph Convolutional Models (GC): Extend circular fingerprints with learnable convolutions over molecular graphs.
- Weave Models: Update atom features using information from all other atoms and their pairwise features.
- Directed Acyclic Graph (DAG) Models: Define directed bonds toward a central atom and propagate features through the directed graph.
- Deep Tensor Neural Networks (DTNN): Use nuclear charges and distance matrices directly, updating atom embeddings based on pairwise physical distances.
- ANI-1: Learns transferable potentials using symmetry function features with atom-type-specific neural networks.
- Message Passing Neural Networks (MPNN): Generalized framework with edge-dependent message functions and set2set readout.
Experimental Protocol
Gaussian process hyperparameter optimization was applied to each dataset-model combination, followed by three independent runs with different random seeds. All results are reported as means with standard deviations. Variable training-size experiments were conducted on Tox21, FreeSolv, and QM7 to study data efficiency.
Key Findings Across Property Categories
Biophysics and Physiology
Graph convolutional and weave models showed strong performance on larger datasets with less overfitting than conventional methods. Graph-based models outperformed multitask networks at 30% training data compared to 90% on Tox21. However, for smaller single-task datasets (under 3,000 samples), kernel SVM and ensemble tree methods were more robust. On highly imbalanced datasets like MUV (0.20% positive rate), graph-based models struggled to control false positives.
Multitask training had a regularizing effect, reducing the gap between train and test scores compared to single-task models. Bypass networks consistently matched or exceeded vanilla multitask networks, confirming that per-task layers add explanatory power.
Physical Chemistry
Graph-based methods (GC, DAG, MPNN, Weave) provided significant improvements over single-task networks for predicting solubility, solvation energy, and lipophilicity. The best models achieved accuracy comparable to ab initio predictions (within 0.5 RMSE for ESOL, within 1.5 kcal/mol for FreeSolv). On FreeSolv, a weave model trained on approximately 200 samples matched the accuracy of alchemical free energy calculations.
Quantum Mechanics
Models incorporating 3D distance information (DTNN, MPNN, KRR with Coulomb matrix) substantially outperformed models using only topological features. DTNN and MPNN covered the best-performing models on 28 of 39 tasks across QM datasets. The choice of physics-aware featurization proved more important than the choice of learning algorithm for these tasks.
Summary of Best Performances
Graph-based models outperformed conventional methods on 11 of 17 datasets. Key results on the test set:
| Dataset | Metric | Best Conventional | Best Graph-Based |
|---|---|---|---|
| QM7 | MAE | KRR (CM): 10.22 | DTNN: 8.75 |
| QM9 | MAE | Multitask (CM): 4.35 | DTNN: 2.35 |
| ESOL | RMSE | XGBoost: 0.99 | MPNN: 0.58 |
| FreeSolv | RMSE | XGBoost: 1.74 | MPNN: 1.15 |
| PCBA | PRC-AUC | Logreg: 0.129 | GC: 0.136 |
| Tox21 | ROC-AUC | KernelSVM: 0.822 | GC: 0.829 |
| HIV | ROC-AUC | KernelSVM: 0.792 | GC: 0.763 |
| BACE | ROC-AUC | RF: 0.867 | Weave: 0.806 |
Conventional methods (KernelSVM, RF) still won on several smaller or scaffold-split datasets (HIV, BACE, MUV, PDBbind, BBBP, SIDER), highlighting that graph-based models are not universally superior, particularly under data scarcity or challenging splits.
Conclusions and Limitations
MoleculeNet demonstrated that learnable representations broadly offer the best performance for molecular machine learning. However, the authors identify several important caveats:
- Data scarcity: Graph-based methods are not robust enough on complex tasks with limited training data.
- Class imbalance: On heavily imbalanced classification datasets, conventional methods such as kernel SVM outperform learnable featurizations with respect to recall of positives.
- Task-specific featurizations: For quantum mechanical and biophysical datasets, incorporating physics-aware features (Coulomb matrix, 3D coordinates) is more important than the choice of learning algorithm.
- Data-driven physical chemistry: On FreeSolv, data-driven methods outperformed ab initio calculations with moderate data, suggesting data-driven approaches will become increasingly important as methods and datasets mature.
The authors express hope that MoleculeNet will stimulate algorithmic development similar to how ImageNet catalyzed breakthroughs in computer vision. Future directions include extending coverage to 3D protein structure prediction, DNA topological modeling, and other areas of molecular science.
Reproducibility Details
Data
All 17 datasets are publicly available and integrated into the DeepChem Python package. Users can load any dataset with a single library call.
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| QM benchmark | QM7/QM7b/QM8/QM9 | 7K-134K compounds | DFT-computed properties from GDB subsets |
| Physical chemistry | ESOL/FreeSolv/Lipophilicity | 643-4,200 compounds | Experimental measurements |
| Biophysics | PCBA/MUV/HIV/PDBbind/BACE | 1.5K-440K compounds | Bioassay and binding data |
| Physiology | BBBP/Tox21/ToxCast/SIDER/ClinTox | 1.4K-8.6K compounds | Toxicity and drug safety data |
Algorithms
All splitting methods (random, scaffold, stratified, time) and featurizations (ECFP, Coulomb matrix, grid, symmetry functions, graph convolutions, weave) are implemented in DeepChem. Hyperparameters were tuned via Gaussian process optimization. Three random seeds were used per experiment.
Models
All 12 models are implemented in DeepChem, built on Scikit-Learn and TensorFlow. No pretrained weights are provided; models are trained from scratch on each dataset.
Evaluation
Metrics include MAE, RMSE, ROC-AUC, and PRC-AUC as specified per dataset. Multi-task datasets report mean metric values across all tasks.
Hardware
The authors used Stanford’s Sherlock and Xstream GPU nodes. Specific GPU types and training times per model are provided in Table S1 of the supplementary material.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| DeepChem | Code | MIT | Open-source library with all datasets, featurizations, and models |
Paper Information
Citation: Wu, Z., Ramsundar, B., Feinberg, E. N., Gomes, J., Geniesse, C., Pappu, A. S., Leswing, K., & Pande, V. (2018). MoleculeNet: a benchmark for molecular machine learning. Chemical Science, 9(2), 513-530. https://doi.org/10.1039/c7sc02664a
@article{wu2018moleculenet,
title={MoleculeNet: a benchmark for molecular machine learning},
author={Wu, Zhenqin and Ramsundar, Bharath and Feinberg, Evan N. and Gomes, Joseph and Geniesse, Caleb and Pappu, Aneesh S. and Leswing, Karl and Pande, Vijay},
journal={Chemical Science},
volume={9},
number={2},
pages={513--530},
year={2018},
publisher={Royal Society of Chemistry},
doi={10.1039/c7sc02664a}
}