The Role of MOSES: A Benchmarking Resource
This is a Resource and Benchmarking paper. It introduces Molecular Sets (MOSES), a platform designed to standardize the training, comparison, and evaluation of molecular generative models. It provides a standardized dataset, a suite of evaluation metrics, and a collection of baseline models to serve as reference points for the field.
Motivation: The Reproducibility Crisis in Generative Chemistry
Generative models are increasingly popular for drug discovery and material design, capable of exploring the vast chemical space ($10^{23}$ to $10^{80}$ compounds) more efficiently than traditional methods. However, the field faces a significant reproducibility crisis:
- Lack of Standardization: There is no consensus on how to properly compare and rank the efficacy of different generative models.
- Inconsistent Metrics: Different papers use different metrics or distinct implementations of the same metrics.
- Data Variance: Models are often trained on different subsets of chemical databases (like ZINC), making direct comparison impossible.
MOSES aims to solve these issues by providing a unified “measuring stick” for distribution learning models in chemistry.
Core Innovation: Standardizing Chemical Distribution Learning
The core contribution is the standardization of the distribution learning definition for molecular generation. Why focus on distribution learning? Rule-based filters enforce strict boundaries like molecular weight limits. Distribution learning complements this by allowing chemists to apply implicit or soft restrictions. This ensures that generated molecules satisfy hard constraints and reflect complex chemical realities defined by the training distribution. These realities include the prevalence of certain substructures and the avoidance of unstable motifs.
MOSES specifically targets distribution learning by providing:
- A Clean, Standardized Dataset: A specific subset of the ZINC Clean Leads collection with rigorous filtering.
- Diverse Metrics: A comprehensive suite of metrics that measure validity alongside novelty, diversity (internal and external), chemical properties (properties distribution), and substructure similarity.
- Open Source Platform: A Python library
molsetsthat decouples the data and evaluation logic from the model implementation, ensuring everyone measures performance exactly the same way.
Experimental Setup and Baseline Generative Models
The authors benchmarked a wide variety of generative models against the MOSES dataset to establish baselines:
- Baselines: Character-level RNN (CharRNN), Variational Autoencoder (VAE), Adversarial Autoencoder (AAE), Junction Tree VAE (JTN-VAE), and LatentGAN.
- Non-Neural Baselines: HMM, n-gram models, and a combinatorial generator (randomly connecting fragments).
- Evaluation: Models were trained on the standard set and evaluated on:
- Validity/Uniqueness: Can the model generate valid, non-duplicate SMILES? Uniqueness is measured at $k = 1{,}000$ and $k = 10{,}000$ samples.
- Filters: What fraction of generated molecules pass the same medicinal chemistry and PAINS filters used for dataset construction?
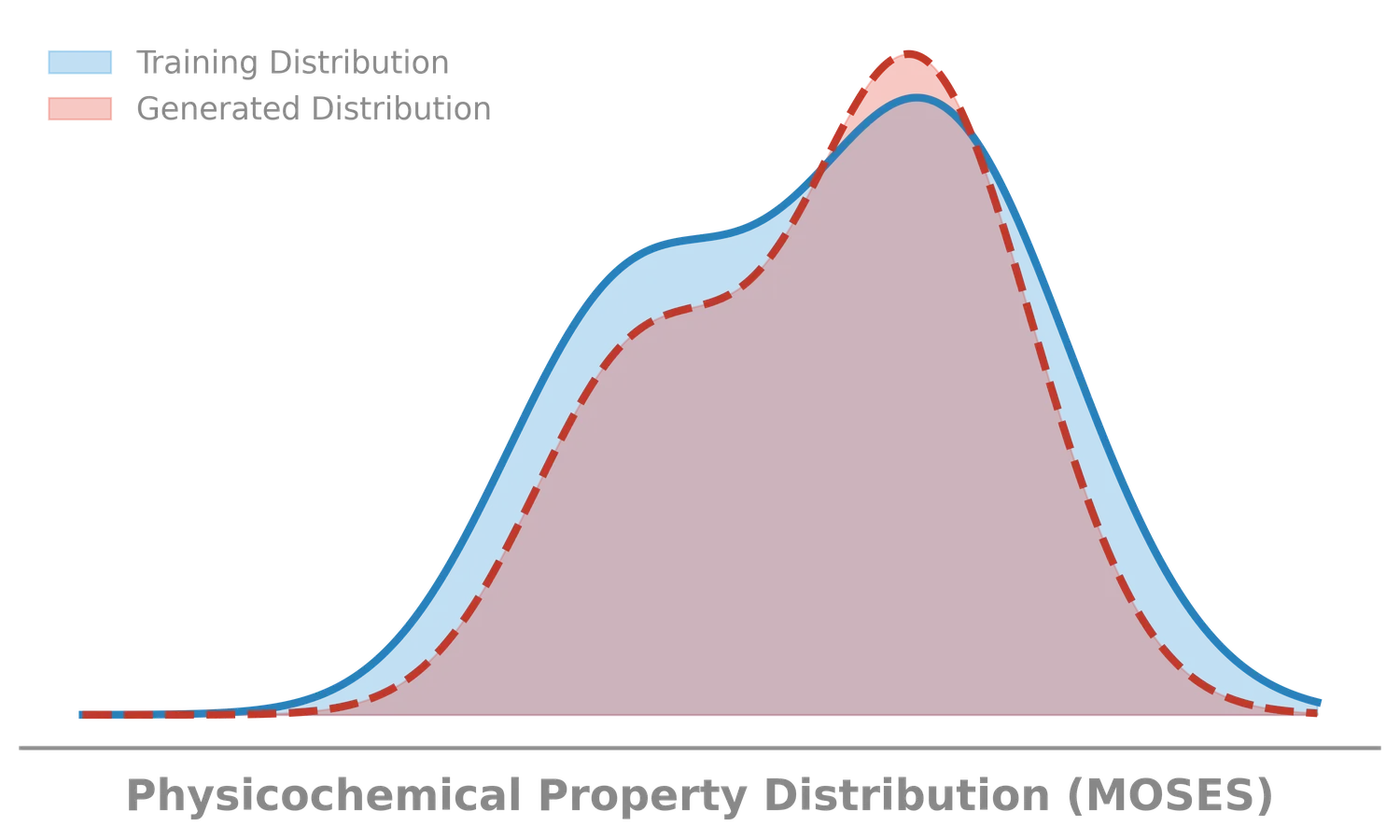
- Feature Distribution: Do generated molecules match the physicochemical properties of the training set? Evaluated using the Wasserstein-1 distance on 1D distributions of:
- LogP: Octanol-water partition coefficient (lipophilicity).
- SA: Synthetic Accessibility score (ease of synthesis).
- QED: Quantitative Estimation of Drug-likeness.
- MW: Molecular Weight.
- Fréchet ChemNet Distance (FCD): Measures similarity in biological/chemical space using the penultimate-layer (second-to-last layer) activations of a pre-trained network (ChemNet).
- Similarity to Nearest Neighbor (SNN): Measures the precision of generation by checking the closest match in the training set (Tanimoto similarity).
Key Findings and Metric Trade-offs
- CharRNN Performance: The simple character-level RNN (CharRNN) outperformed more complex models (like VAEs and GANs) on many metrics, achieving the best FCD scores ($0.073$).
- Metric Trade-offs: No single metric captures “quality.”
- The Combinatorial Generator achieved 100% validity and high diversity. It struggled with distribution learning metrics (FCD), indicating it explores chemical space broadly without capturing natural distributions.
- VAEs often achieve high Similarity to Nearest Neighbor (SNN) while exhibiting low novelty. The authors suggest this pattern may indicate overfitting to training set prototypes, though they treat this as a hypothesis rather than a proven mechanism.
- Implicit Constraints: A major finding was that neural models successfully learned implicit chemical rules (like avoiding PAINS structures) purely from the data distribution.
- Recommendation: The authors suggest using FCD/Test for general model ranking, while emphasizing the importance of checking specific metrics (validity, diversity) to diagnose model failure modes.
- Limitations of the Benchmark: MOSES focuses on distribution learning and uses FCD as a primary ranking metric. As the authors note, FCD captures multiple aspects of other metrics in a single number but does not give insights into specific issues, so more interpretable metrics are necessary for thorough investigation. The benchmark evaluates only 1D (SMILES) and 2D molecular features, without assessing 3D conformational properties.
Reproducibility Details
Data
The benchmark uses a curated subset of the ZINC Clean Leads collection.
- Source Size: ~4.6M molecules (4,591,276 after initial extraction).
- Final Size: 1,936,962 molecules.
- Splits: Train (1,584,664), Test (176,075), Scaffold Test (176,226).
- Scaffold Test Split: This split is crucial for distinct generalization testing. It contains molecules whose Bemis-Murcko scaffolds are completely absent from the training and test sets. Evaluating on this split strictly tests a model’s ability to generate novel chemical structures (generalization).
- Filters Applied:
- Molecular weight: 250 to 350 Da
- Rotatable bonds: $\leq 7$
- XlogP: $\leq 3.5$
- Atom types: C, N, S, O, F, Cl, Br, H
- No charged atoms or cycles > 8 atoms
- Medicinal Chemistry Filters (MCF) and PAINS filters applied.
Evaluation Metrics
MOSES introduces a standard suite of metrics. Key definitions:
- Validity: Fraction of valid SMILES strings (via RDKit).
- Unique@k: Fraction of unique molecules in the first $k$ valid samples ($k = 1{,}000$ and $k = 10{,}000$).
- Filters: Fraction of generated molecules passing the MCF and PAINS filters used during dataset construction. High scores here indicate the model learned implicit chemical validity constraints from the data distribution.
- Novelty: Fraction of generated molecules not present in the training set.
- Internal Diversity (IntDiv): Average Tanimoto distance between generated molecules ($G$), useful for detecting mode collapse: $$ \text{IntDiv}_p(G) = 1 - \sqrt[p]{\frac{1}{|G|^2} \sum_{m_1, m_2 \in G} T(m_1, m_2)^p} $$
- Fragment Similarity (Frag): Cosine similarity of fragment frequency vectors (BRICS decomposition) between generated and test sets.
- Scaffold Similarity (Scaff): Cosine similarity of Bemis-Murcko scaffold frequency vectors between sets. Measures how well the model captures higher-level structural motifs.
- Similarity to Nearest Neighbor (SNN): The average Tanimoto similarity between a generated molecule’s fingerprint and its nearest neighbor in the reference set. This serves as a measure of precision; high SNN suggests the model produces molecules very similar to the training distribution, potentially indicating memorization if novelty is low. $$ \text{SNN}(G, R) = \frac{1}{|G|} \sum_{m_G \in G} \max_{m_R \in R} T(m_G, m_R) $$
- Fréchet ChemNet Distance (FCD): Fréchet distance between the Gaussian approximations (mean and covariance) of penultimate-layer activations from ChemNet. This measures how close the distribution of generated molecules is to the real distribution in chemical/biological space. The authors note that FCD correlates with other metrics. For example, if the generated structures are not diverse enough or the model produces too many duplicates, FCD will decrease because the variance is smaller. The authors suggest using FCD for hyperparameter tuning and final model selection. $$ \text{FCD}(G, R) = |\mu_G - \mu_R|^2 + \text{Tr}(\Sigma_G + \Sigma_R - 2(\Sigma_G \Sigma_R)^{1/2}) $$
- Properties Distribution (Wasserstein-1): The 1D Wasserstein-1 distance between the distributions of molecular properties (MW, LogP, SA, QED) in the generated and test sets.
Models & Baselines
The paper selects baselines to represent different theoretical approaches to distribution learning:
- Explicit Density Models: Models where the probability mass function $P(x)$ can be computed analytically.
- N-gram: Simple statistical models. They failed to generate valid molecules reliably due to limited long-range dependency modeling.
- Implicit Density Models: Models that sample from the distribution without explicitly computing $P(x)$.
- VAE/AAE: Optimizes a lower bound on the log-likelihood (ELBO) or uses adversarial training.
- GANs (LatentGAN): Directly minimizes the distance between real and generated distributions via a discriminator.
Models are also distinguished by their data representation:
- String-based (SMILES): Models like CharRNN, VAE, and AAE treat molecules as SMILES strings. SMILES encodes a molecular graph by traversing a spanning tree in depth-first order, storing atom and edge tokens.
- Graph-based: JTN-VAE operates directly on molecular subgraphs (junction tree), ensuring chemical validity by construction but often requiring more complex training.
Key baselines implemented in PyTorch (hyperparameters are detailed in Supplementary Information 3 of the original paper):
- CharRNN: LSTM-based sequence model (3 layers, 768 hidden units). Trained with Adam ($lr = 10^{-3}$, batch size 64, 80 epochs, learning rate halved every 10 epochs).
- VAE: Encoder-decoder architectures (bidirectional GRU encoder, 3-layer GRU decoder with 512 hidden units) with KL regularization.
- AAE: Encoder (single layer bidirectional LSTM with 512 units) and decoder (2-layer LSTM with 512 units) initialized with adversarial formulation.
- LatentGAN: GAN (5-layer fully connected generator) trained on the latent space of a pre-trained heteroencoder.
- JTN-VAE: Tree-structured graph generation.
Code & Hardware Requirements
- Code Repository: Available at github.com/molecularsets/moses as well as the PyPI library
molsets. The platform provides standard scripts (scripts/run.pyto evaluate models end-to-end, andscripts/run_all_models.shfor multi-seed evaluations). - Hardware: The repository supports GPU acceleration via
nvidia-docker(defaulting to 10GB shared memory). However, specific training times and exact GPU models used by the authors for the baselines are not formally documented in the source text. - Model Weights: Pre-trained model checkpoints are not natively pre-packaged as standalone downloads; practitioners are expected to re-train the default baselines using the provided scripts.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| molecularsets/moses | Code | MIT | Official benchmark platform with baseline models and evaluation metrics |
| molsets (PyPI) | Code | MIT | pip-installable package for dataset access and metric computation |
| ZINC Clean Leads subset | Dataset | See ZINC terms | Curated dataset of 1,936,962 molecules distributed via the repository |
Paper Information
Citation: Polykovskiy, D., Zhebrak, A., Sanchez-Lengeling, B., Golovanov, S., Tatanov, O., Belyaev, S., Kurbanov, R., Artamonov, A., Aladinskiy, V., Veselov, M., Kadurin, A., Johansson, S., Chen, H., Nikolenko, S., Aspuru-Guzik, A., and Zhavoronkov, A. (2020). Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Frontiers in Pharmacology, 11, 565644. https://doi.org/10.3389/fphar.2020.565644
Publication: Frontiers in Pharmacology, 2020
@article{polykovskiy2020moses,
title={Molecular Sets (MOSES): A benchmarking platform for molecular generation models},
author={Polykovskiy, Daniil and Zhebrak, Alexander and Sanchez-Lengeling, Benjamin and Golovanov, Sergey and Tatanov, Oktai and Belyaev, Stanislav and Kurbanov, Rauf and Artamonov, Aleksey and Aladinskiy, Vladimir and Veselov, Mark and Kadurin, Artur and Johansson, Simon and Chen, Hongming and Nikolenko, Sergey and Aspuru-Guzik, Al{\'a}n and Zhavoronkov, Alex},
journal={Frontiers in pharmacology},
volume={11},
pages={565644},
year={2020},
publisher={Frontiers},
doi={10.3389/fphar.2020.565644}
}