A Graph-Based Approach to Molecular Optimization
This is a Method paper that introduces two graph-based approaches for exploring chemical space: a genetic algorithm (GB-GA) and a generative model combined with Monte Carlo tree search (GB-GM-MCTS). The primary contribution is demonstrating that these non-ML, graph-based methods can match or exceed the performance of contemporary ML-based generative models for molecular property optimization, while being several orders of magnitude faster. The paper provides open-source implementations built on the RDKit cheminformatics package. The two approaches explore chemical space using direct graph manipulations rather than string-based representations like SMILES.
Why Compare Simple Baselines to ML Generative Models?
By 2018, several ML-based generative models for molecules had been published, including VAEs, RNNs, and graph convolutional policy networks. However, these models were rarely compared against traditional optimization approaches such as genetic algorithms. Jensen identifies this gap explicitly: while ML generative model performance had been impressive, the lack of comparison to simpler baselines made it difficult to assess whether the complexity of ML approaches was justified.
A practical barrier to such comparisons was the absence of free, open-source GA implementations for molecular optimization (the existing ACSESS algorithm required proprietary OpenEye toolkits). This paper fills that gap by providing RDKit-based implementations of both the GB-GA and GB-GM-MCTS.
Graph-Based Crossovers, Mutations, and Monte Carlo Tree Search
GB-GA: Crossovers and Mutations on Molecular Graphs
The GB-GA operates directly on molecular graph representations (not string representations like SMILES). It combines ideas from Brown et al. (2004) and the ACSESS algorithm of Virshup et al. (2013).
Crossovers can occur at two types of positions with equal probability:
- Non-ring bonds: a molecule is cut at a non-ring bond, and fragments from two parent molecules are recombined
- Ring bonds: adjacent bonds or bonds separated by one bond are cut, and fragments are mated using single or double bonds
Mutations include seven operation types, each with specified probabilities:
- Append atom (15%): adds an atom with a single, double, or triple bond
- Insert atom (15%): inserts an atom into an existing bond
- Delete atom (14%): removes an atom, reconnecting neighbors
- Change atom type (14%): swaps element identity (C, N, O, F, S, Cl, Br)
- Change bond order (14%): toggles between single, double, and triple bonds
- Delete ring bond (14%): opens a ring
- Add ring bond (14%): closes a new ring
Molecules with macrocycles (seven or more atoms), allene centers in rings, fewer than five heavy atoms, incorrect valences, or more non-H atoms than the target size are discarded. The target size is sampled from a normal distribution with mean 39.15 and standard deviation 3.50 non-H atoms, calibrated to match the molecules found by Yang et al. (2017).
GB-GM-MCTS: A Probabilistic Growth Model with Tree Search
The GB-GM grows molecules one atom at a time, with the choice of bond order and atom type determined probabilistically from a bonding analysis of a reference dataset (the first 1000 molecules from ZINC). Since 63% of atoms in the reference set are ring atoms, ring-creation or ring-insertion mutations are chosen 63% of the time.
The generative model is combined with a Monte Carlo tree search where:
- Each node corresponds to an atom addition step
- Leaf parallelization uses a maximum of 25 leaf nodes
- The exploration factor is $1 / \sqrt{2}$
- Rollout terminates if the molecule exceeds the target size
- The reward function returns 1 if the predicted $J(\mathbf{m})$ value exceeds the largest value found so far, and 0 otherwise
The Penalized logP Objective
Both methods optimize the penalized logP score $J(\mathbf{m})$:
$$ J(\mathbf{m}) = \log P(\mathbf{m}) - \text{SA}(\mathbf{m}) - \text{RingPenalty}(\mathbf{m}) $$
where $\log P(\mathbf{m})$ is the octanol-water partition coefficient predicted by RDKit, $\text{SA}(\mathbf{m})$ is a synthetic accessibility score, and $\text{RingPenalty}(\mathbf{m})$ penalizes unrealistically large rings by reducing the score by $\text{RingSize} - 6$ for each oversized ring. Each property is normalized to zero mean and unit standard deviation across the ZINC dataset.
Experimental Setup and Comparisons to ML Methods
GB-GA Experiments
Ten GA simulations were performed with a population size of 20 over 50 generations (1000 $J(\mathbf{m})$ evaluations per run). The initial mating pool was 20 random molecules from the first 1000 molecules in ZINC. Two mutation rates were tested: 50% and 1%.
GB-GM-MCTS Experiments
Ten simulations used ethane as a seed molecule with 1000 tree traversals per run. Additional experiments used 5000 traversals and an adjusted probability of generating $\text{C}=\text{C}-\text{C}$ ring patterns (increased from 62% to 80%).
Baselines
Results were compared to those compiled by Yang et al. (2017):
- ChemTS (RNN + MCTS)
- RNN with and without Bayesian optimization
- Continuous VAE (CVAE)
- Grammar VAE (GVAE)
- Graph convolutional policy network (GCPN, from You et al. 2018)
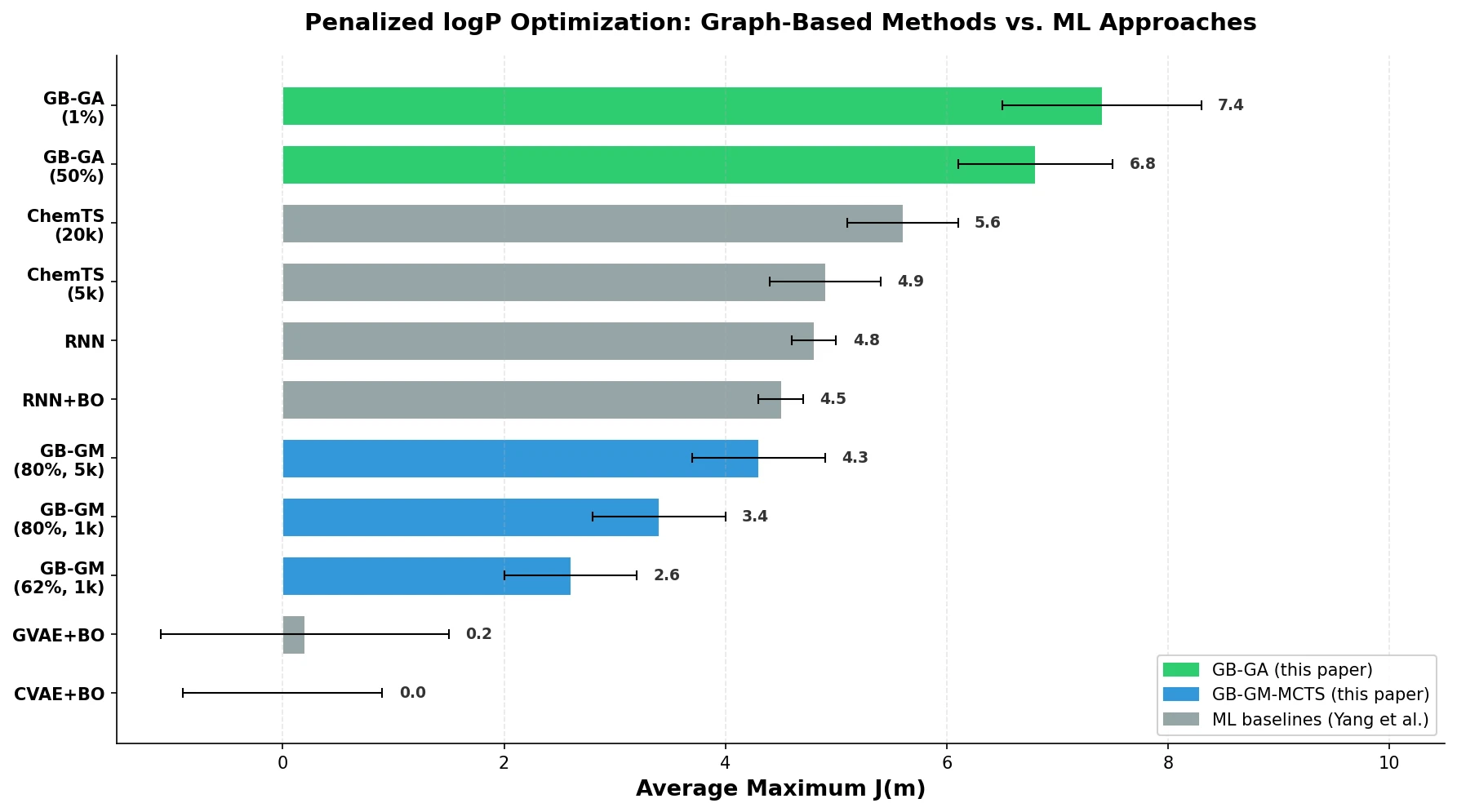
Key Results
| Method | Average $J(\mathbf{m})$ | Molecules Evaluated | CPU Time |
|---|---|---|---|
| GB-GA (50% mutation) | 6.8 +/- 0.7 | 1000 | 30 seconds |
| GB-GA (1% mutation) | 7.4 +/- 0.9 | 1000 | 30 seconds |
| GB-GM-MCTS (62%) | 2.6 +/- 0.6 | 1000 | 90 seconds |
| GB-GM-MCTS (80%) | 3.4 +/- 0.6 | 1000 | 90 seconds |
| GB-GM-MCTS (80%) | 4.3 +/- 0.6 | 5000 | 9 minutes |
| ChemTS | 4.9 +/- 0.5 | ~5000 | 2 hours |
| ChemTS | 5.6 +/- 0.5 | ~20000 | 8 hours |
| RNN + BO | 4.5 +/- 0.2 | ~4000 | 8 hours |
| Only RNN | 4.8 +/- 0.2 | ~20000 | 8 hours |
| CVAE + BO | 0.0 +/- 0.9 | ~100 | 8 hours |
| GVAE + BO | 0.2 +/- 1.3 | ~1000 | 8 hours |
The GB-GA with 1% mutation rate achieved an average maximum $J(\mathbf{m})$ of 7.4, which is 1.8 units higher than the best ML result (ChemTS at 5.6) while using 20x fewer evaluations and completing in 30 seconds versus 8 hours. The two highest-scoring individual molecules found by GB-GA had $J(\mathbf{m})$ scores of 8.8 and 8.5, exceeding the 7.8-8.0 range found by the GCPN approach. These molecules bore little resemblance to the initial mating pool (Tanimoto similarities of 0.27 and 0.12 to the most similar ZINC molecules), indicating that the GA traversed a large distance in chemical space in just 50 generations.
The GB-GM-MCTS performed below ChemTS at equal evaluations (4.3 vs. 4.9 at 5000 evaluations) but was several orders of magnitude faster (9 minutes vs. 2 hours). The MCTS approach tended to extract the dominant hydrophobic structural motif (benzene rings) from the training set, making it more dependent on training set composition than the GA.
Simple Methods Set a High Bar for Molecular Optimization
The central finding is that a simple graph-based genetic algorithm outperforms all tested ML-based generative models on penalized logP optimization, both in terms of solution quality and computational efficiency. The GB-GA achieves higher $J(\mathbf{m})$ scores with 1000 evaluations in 30 seconds than ML methods achieve with 20,000 evaluations over 8 hours.
Several additional observations emerge:
- Chemical space traversal: The GB-GA can reach high-scoring molecules that are structurally distant from the starting population, with Tanimoto similarity as low as 0.12 to the nearest ZINC molecule.
- Mutation rate matters: A 1% mutation rate outperformed a 50% rate (7.4 vs. 6.8), suggesting that preserving more parental structure during crossover is beneficial for this objective.
- Training set dependence: The GB-GM-MCTS is more sensitive to training set composition than the GA. Its preference for benzene-ring-containing molecules (the dominant ZINC motif) limits its ability to discover alternative structural solutions like the long aliphatic chains favored by the GA.
- Generalizability caveat: Jensen explicitly notes that these comparisons cover only one property (penalized logP) and that similar comparisons for other properties are needed before drawing general conclusions.
The paper’s influence has been substantial: it helped establish the expectation that new molecular generative models should be benchmarked against genetic algorithm baselines, a position subsequently reinforced by Brown et al. (2019) in GuacaMol and by Tripp and Hernandez-Lobato (2023).
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Initial mating pool / reference set | ZINC (subset) | First 1000 molecules | Same subset used in previous studies (Gomez-Bombarelli et al., Yang et al.) |
| Target molecule size | Derived from Yang et al. results | 20 molecules | Mean 39.15, SD 3.50 non-H atoms |
Algorithms
- GB-GA: Population size 20, 50 generations, mutation rates of 1% and 50% tested. Crossovers at ring and non-ring bonds with equal probability. Seven mutation types with specified probabilities. Molecules selected from mating pool based on normalized logP scores.
- GB-GM: Atom-by-atom growth using probabilistic rules derived from ZINC bonding analysis. Ring creation probability 63% (matching ZINC), with 80% variant also tested. Seed molecule: ethane.
- MCTS: Modified from haroldsultan/MCTS Python implementation. Leaf parallelization with max 25 leaf nodes. Exploration factor $1/\sqrt{2}$. Binary reward function (1 if new best, 0 otherwise).
- Property calculation: logP, SA score, and ring penalty all computed via RDKit. Each property normalized to zero mean and unit standard deviation across ZINC.
Models
No neural network models are used. The GB-GA and GB-GM are purely algorithmic approaches parameterized by bonding statistics from the ZINC dataset.
Evaluation
| Metric | GB-GA (1%) | Best ML (ChemTS) | Notes |
|---|---|---|---|
| Average max $J(\mathbf{m})$ | 7.4 +/- 0.9 | 5.6 +/- 0.5 | Over 10 runs |
| Single best $J(\mathbf{m})$ | 8.8 | ~8.0 (GCPN) | GB-GA vs. You et al. |
| Evaluations per run | 1000 | ~20,000 | 20x fewer for GB-GA |
| CPU time per run | 30 seconds | 8 hours | ~960x faster |
Hardware
All GB-GA and GB-GM experiments were run on a laptop. No GPU required. The GB-GA completes in 30 seconds per run and the GB-GM-MCTS in 90 seconds (1000 traversals) to 9 minutes (5000 traversals).
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| GB-GA (v0.0) | Code | Not specified | Graph-based genetic algorithm, RDKit dependency only |
| GB-GM (v0.0) | Code | Not specified | Graph-based generative model + MCTS, RDKit dependency only |
Paper Information
Citation: Jensen, J. H. (2019). A graph-based genetic algorithm and generative model/Monte Carlo tree search for the exploration of chemical space. Chemical Science, 10(12), 3567-3572. https://doi.org/10.1039/c8sc05372c
Publication: Chemical Science (Royal Society of Chemistry), 2019
Additional Resources:
Citation
@article{jensen2019graph,
title={A graph-based genetic algorithm and generative model/Monte Carlo tree search for the exploration of chemical space},
author={Jensen, Jan H.},
journal={Chemical Science},
volume={10},
number={12},
pages={3567--3572},
year={2019},
publisher={Royal Society of Chemistry},
doi={10.1039/c8sc05372c}
}