A Unified Evaluation Metric for Molecular Generation
This is a Method paper that introduces the Frechet ChemNet Distance (FCD), a single scalar metric for evaluating generative models that produce molecules for drug discovery. FCD adapts the Frechet Inception Distance (FID) from image generation to the molecular domain. By comparing distributions of learned representations from a drug-activity prediction network (ChemNet), FCD simultaneously captures whether generated molecules are chemically valid, biologically relevant, and structurally diverse.
Inconsistent Evaluation of Molecular Generative Models
At the time of this work (2018), deep generative models for molecules were proliferating: RNNs combined with variational autoencoders, reinforcement learning, and GANs all produced SMILES strings representing novel molecules. The evaluation landscape was fragmented. Different papers reported different metrics: percentage of valid SMILES, mean logP, druglikeness, synthetic accessibility (SA) scores, or internal diversity via Tanimoto distance.
This inconsistency created several problems. First, method comparison across publications was difficult because no common metric existed. Second, simple metrics like “fraction of valid SMILES” could be trivially maximized by generating short, simple molecules (e.g., “CC” or “CCC”). Third, individual property metrics (logP, druglikeness) each captured only one dimension of quality. A model could score well on logP but produce molecules that were not diverse or not biologically meaningful.
The authors argued that a good metric should capture three properties simultaneously: (1) chemical validity and similarity to real drug-like molecules, (2) biological relevance, and (3) diversity within the generated set.
Core Innovation: Frechet Distance over ChemNet Activations
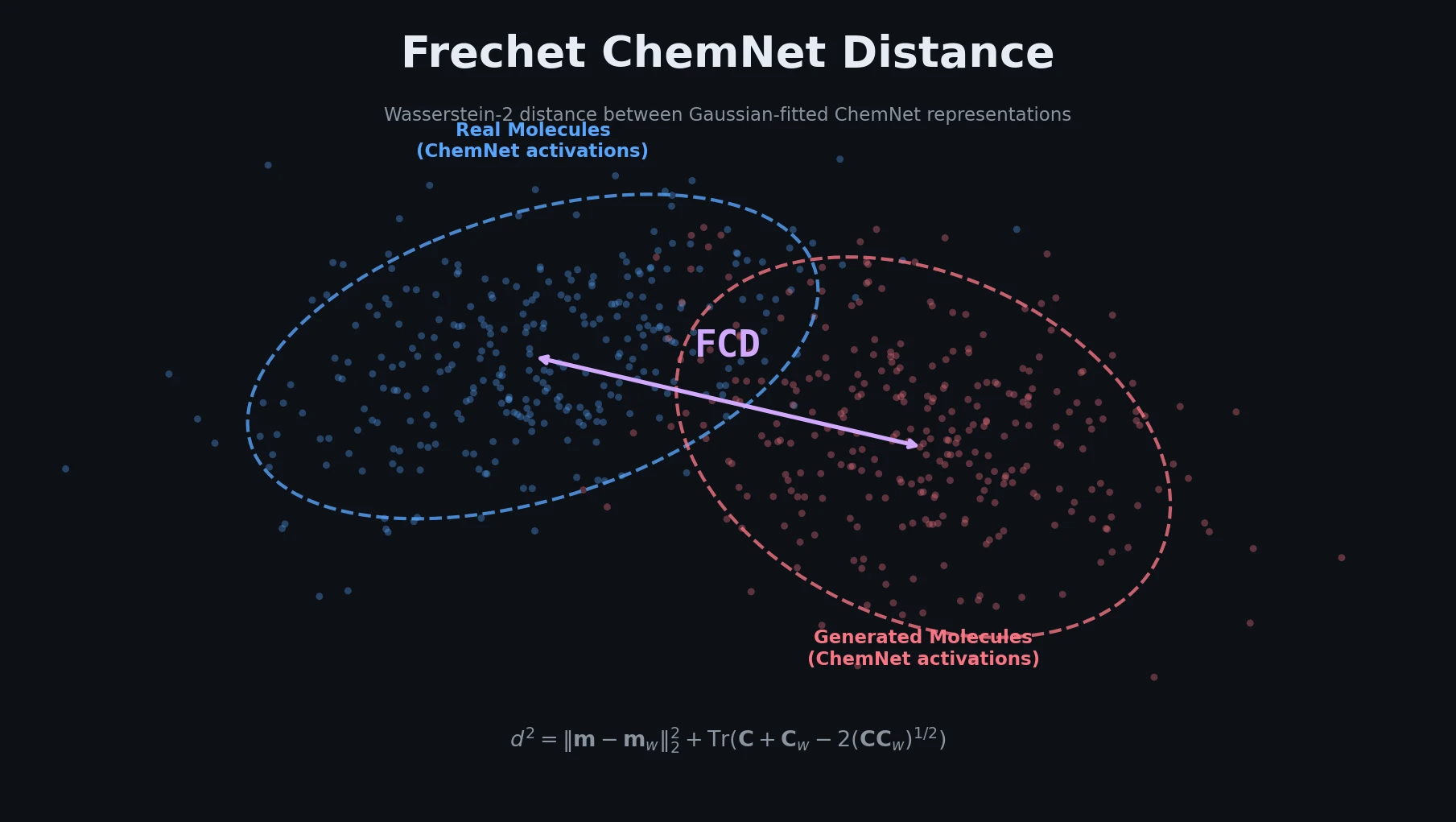
The key insight is to use a neural network trained on biological activity prediction as a feature extractor for molecules, then compare distributions of these features using the Frechet (Wasserstein-2) distance.
ChemNet Architecture
ChemNet is a multi-task neural network trained to predict bioactivities across approximately 6,000 assays from three major drug discovery databases (ChEMBL, ZINC, PubChem). The architecture processes one-hot encoded SMILES strings through:
- Two 1D convolutional layers with SELU activations
- A max-pooling layer
- Two stacked LSTM layers
- A fully connected output layer
The penultimate layer (the second LSTM’s hidden state after processing the full input sequence) serves as the molecular representation. Because ChemNet was trained to predict drug activities, its internal representations encode both chemical structure (from the input side) and biological function (from the output side).
The FCD Formula
Given a set of real molecules and a set of generated molecules, FCD is computed as follows:
- Pass each molecule (as a SMILES string) through ChemNet and extract penultimate-layer activations.
- Fit a multivariate Gaussian to each set by computing the mean $\mathbf{m}$ and covariance $\mathbf{C}$ for the generated set, and mean $\mathbf{m}_w$ and covariance $\mathbf{C}_w$ for the real set.
- Compute the squared Frechet distance:
$$ d^{2}\left((\mathbf{m}, \mathbf{C}), (\mathbf{m}_w, \mathbf{C}_w)\right) = |\mathbf{m} - \mathbf{m}_w|_2^{2} + \mathrm{Tr}\left(\mathbf{C} + \mathbf{C}_w - 2(\mathbf{C}\mathbf{C}_w)^{1/2}\right) $$
The Gaussian assumption is justified by the maximum entropy principle: the Gaussian is the maximum-entropy distribution for given mean and covariance. A lower FCD indicates that the generated distribution is closer to the real distribution.
Why Not Just Fingerprints?
The authors also define a Frechet Fingerprint Distance (FFD) that replaces ChemNet activations with 2048-bit ECFP_4 fingerprints. FFD captures chemical structure but not biological function. The experimental comparison shows that FCD produces more distinct separations between biased and unbiased molecule sets, particularly for biologically meaningful biases.
Detecting Flaws in Generative Models
The experiments evaluate whether FCD can detect specific failure modes in generative models. The authors simulate five types of biased generators by selecting molecules from real databases that exhibit particular properties, then compare FCD against individual metrics (logP, druglikeness, SA score, internal diversity) and FFD.
Simulated Bias Experiments
All experiments use 5,000 molecules drawn 5 times each. The reference distribution is 200,000 randomly drawn real molecules not used for ChemNet training.
| Bias Type | logP | Druglikeness | SA Score | Int. Diversity | FFD | FCD |
|---|---|---|---|---|---|---|
| Low druglikeness (<5th pct) | - | Detects | - | - | Detects | Detects |
| High logP (>95th pct) | Detects | Detects | - | - | Detects | Detects |
| Low SA score (<5th pct) | - | Partial | - | Partial | Detects | Detects |
| Mode collapse (cluster) | - | - | - | Detects | Detects | Detects |
| Kinase inhibitors (PLK1) | - | - | - | - | Detects | Detects |
FCD is the only metric that detects all five bias types. The biological bias test (kinase inhibitors for PLK1-PBD from PubChem AID 720504) is particularly notable: only FFD and FCD detect this bias, and FCD provides a more distinct separation. This validates the hypothesis that incorporating biological information through ChemNet activations improves evaluation beyond purely chemical descriptors.
Sample Size Requirements
The authors tested FCD convergence with varying sample sizes (5 to 300,000 molecules). Mean FCD values for samples drawn from the real distribution:
| Sample Size | Mean FCD | Std Dev |
|---|---|---|
| 5 | 76.46 | 5.03 |
| 50 | 31.86 | 0.75 |
| 500 | 4.41 | 0.03 |
| 5,000 | 0.42 | 0.01 |
| 50,000 | 0.05 | 0.00 |
| 300,000 | 0.02 | 0.00 |
A sample size of 5,000 molecules is sufficient for reliable estimation, with the mean FCD approaching zero and negligible variance.
Benchmarking Published Generative Models
The authors computed FCD for several published generative methods:
| Method | FCD | Notes |
|---|---|---|
| Random real molecules | 0.22 | Baseline (near zero as expected) |
| Segler et al. (LSTM) | 1.62 | Trained to approximate full ChEMBL distribution |
| DRD2-targeted methods | 24.14 to 47.85 | Olivecrona, RL, and ORGAN agents |
| Rule-based baseline | 58.76 | Random concatenation of C, N, O atoms |
The ranking matches expectations. The Segler model, trained to approximate the overall molecule distribution, achieves the lowest FCD (1.62). Models optimized for a specific target (DRD2), including the Olivecrona RL agents, the RL method by Benhenda, and ORGAN, produce higher FCD values (24.14 to 47.85) against the general distribution. More training iterations push these models further from the general distribution, as they become increasingly DRD2-specific. The canonical and reduced Olivecrona agents learn similar chemical spaces, consistent with the original authors’ conclusions. The rule-based system scores worst (58.76), confirming FCD as a meaningful quality metric.
Conclusions and Impact
FCD provides a single metric that unifies the evaluation of chemical validity, biological relevance, and diversity for molecular generative models. Its main advantages are:
- It captures multiple quality dimensions in one score, simplifying method comparison.
- It detects biases that no single existing metric can catch alone.
- It requires only SMILES strings as input, making it applicable to any generative method (including graph-based approaches via SMILES conversion).
- It incorporates biological information through ChemNet, distinguishing it from purely chemical metrics like FFD.
Limitations: The metric depends on the ChemNet model, which was trained on a specific set of bioactivity assays. Molecules outside the training distribution of ChemNet may not be well-represented. The Gaussian assumption for the activation distributions may not hold perfectly. FCD measures distance to a reference set, so it evaluates how well a generator approximates a given distribution rather than the absolute quality of individual molecules. When using FCD for targeted generation (e.g., molecules active against a specific protein), the reference set should be chosen accordingly, not the general drug-like molecule distribution.
FCD has since become a standard evaluation metric in the molecular generation community, adopted by benchmarking platforms like MOSES and GuacaMol.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| ChemNet training | ChEMBL, ZINC, PubChem | ~6,000 assays | Two-thirds for training, one-third for testing |
| Reference distribution | Combined databases | 200,000 molecules | Excluded from ChemNet training |
| Bias simulations | Subsets of combined databases | 5,000 per experiment | 5 repetitions each |
Algorithms
- ChemNet: 2x 1D-conv (SELU), max-pool, 2x stacked LSTM, FC output
- FCD: Squared Frechet distance between Gaussian-fitted ChemNet penultimate-layer activations
- FFD: Same as FCD but using 2048-bit ECFP_4 fingerprints instead of ChemNet activations
- Molecular property calculations: RDKit (logP, druglikeness, SA score, Morgan fingerprints with radius 2)
Evaluation
| Metric | Description |
|---|---|
| FCD | Frechet distance over ChemNet activations (lower = closer to reference) |
| FFD | Frechet distance over ECFP_4 fingerprints |
| logP | Mean partition coefficient |
| Druglikeness | Geometric mean of desired molecular properties (QED) |
| SA Score | Synthetic accessibility score |
| Internal Diversity | Tanimoto distance within generated set |
Hardware
Hardware specifications are not provided in the paper.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| FCD Implementation | Code | LGPL-3.0 | Official Python implementation; requires only SMILES input |
Paper Information
Citation: Preuer, K., Renz, P., Unterthiner, T., Hochreiter, S., & Klambauer, G. (2018). Fréchet ChemNet Distance: A Metric for Generative Models for Molecules in Drug Discovery. Journal of Chemical Information and Modeling, 58(9), 1736-1741.
@article{preuer2018frechet,
title={Fr{\'e}chet ChemNet Distance: A Metric for Generative Models for Molecules in Drug Discovery},
author={Preuer, Kristina and Renz, Philipp and Unterthiner, Thomas and Hochreiter, Sepp and Klambauer, G{\"u}nter},
journal={Journal of Chemical Information and Modeling},
volume={58},
number={9},
pages={1736--1741},
year={2018},
doi={10.1021/acs.jcim.8b00234},
publisher={American Chemical Society}
}