A Three-Part Resource for Docking-Based ML Benchmarks
DOCKSTRING is a Resource paper that delivers three integrated components for benchmarking machine learning models in drug discovery using molecular docking. The primary contributions are: (1) an open-source Python package wrapping AutoDock Vina for deterministic docking from SMILES strings, (2) a dataset of over 15 million docking scores and poses covering 260,000+ molecules docked against 58 medically relevant protein targets, and (3) a suite of benchmark tasks spanning regression, virtual screening, and de novo molecular design. The paper additionally provides baseline results across classical and deep learning methods.
Why Existing Molecular Benchmarks Fall Short
ML methods for drug discovery are frequently evaluated using simple physicochemical properties such as penalized logP or QED (quantitative estimate of druglikeness). These properties are computationally cheap and easy to optimize, but they do not depend on the interaction between a candidate compound and a protein target. As a result, strong performance on logP or QED benchmarks does not necessarily translate to strong performance on real drug design tasks.
Molecular docking offers a more realistic evaluation objective because docking scores depend on the 3D structure of the ligand-target complex. Docking is routinely used by medicinal chemists to estimate binding affinities during hit discovery and lead optimization. Several prior efforts attempted to bring docking into ML benchmarking, but each had limitations:
- VirtualFlow and DockStream require manually prepared target files and domain expertise.
- TDC and Cieplinski et al. provide SMILES-to-score wrappers but lack proper ligand protonation and randomness control, and cover very few targets (one and four, respectively).
- DUD-E is easily overfit by ML models that memorize actives vs. decoys.
- GuacaMol and MOSES rely on physicochemical properties or similarity functions that miss 3D structural subtleties.
- MoleculeNet compiles experimental datasets but does not support on-the-fly label computation needed for transfer learning or de novo design.
DOCKSTRING addresses all of these gaps: it standardizes the docking procedure, automates ligand and target preparation, controls randomness for reproducibility, and provides a large, diverse target set.
Core Innovation: Standardized End-to-End Docking Pipeline
The key innovation is a fully automated, deterministic docking pipeline that produces reproducible scores from a SMILES string in four lines of Python code. The pipeline consists of three stages:
Target Preparation. 57 of the 58 protein targets originate from the Directory of Useful Decoys Enhanced (DUD-E). PDB files are standardized with Open Babel, polar hydrogens are added, and conversion to PDBQT format is performed with AutoDock Tools. Search boxes are derived from crystallographic ligands with 12.5 A padding and a minimum side length of 30 A. The 58th target (DRD2, dopamine receptor D2) was prepared separately following the same protocol.
Ligand Preparation. Ligands are protonated at pH 7.4 with Open Babel, embedded into 3D conformations using the ETKDG algorithm in RDKit, refined with the MMFF94 force field, and assigned Gasteiger partial charges. Stereochemistry of determined stereocenters is maintained, while undetermined stereocenters are assigned randomly but consistently across runs.
Docking. AutoDock Vina runs with default exhaustiveness (8), up to 9 binding modes, and an energy range of 3 kcal/mol. The authors verified that fixing the random seed yields docking score variance of less than 0.1 kcal/mol across runs, making the pipeline fully deterministic.
The three de novo design objective functions incorporate a QED penalty to enforce druglikeness:
$$ f_{\text{F2}}(l) = s(l, \text{F2}) + 10(1 - \text{QED}(l)) $$
$$ f_{\text{PPAR}}(l) = \max_{t \in \text{PPAR}} s(l, t) + 10(1 - \text{QED}(l)) $$
$$ f_{\text{JAK2}}(l) = s(l, \text{JAK2}) - \min(s(l, \text{LCK}), -8.1) + 10(1 - \text{QED}(l)) $$
The F2 task optimizes binding to a single protease. The Promiscuous PPAR task requires strong binding to three nuclear receptors simultaneously. The Selective JAK2 task is adversarial, requiring strong JAK2 binding while avoiding LCK binding (two kinases with a score correlation of 0.80).
Experimental Setup: Regression, Virtual Screening, and De Novo Design
Dataset Construction
The dataset combines molecules from ExCAPE-DB (which curates PubChem and ChEMBL bioactivity assays). The authors selected all molecules with active labels against targets having at least 1,000 experimental actives, plus 150,000 inactive-only molecules. After discarding 1.8% of molecules that failed ligand preparation, the final dataset contains 260,155 compounds docked against 58 targets, producing over 15 million docking scores and poses. The dataset required over 500,000 CPU hours to generate.
Cluster analysis using DBSCAN (Jaccard distance threshold of 0.25 on RDKit fingerprints) found 52,000 clusters, and Bemis-Murcko scaffold decomposition identified 102,000 scaffolds, confirming high molecular diversity. Train/test splitting follows cluster labels to prevent data leakage.
Regression Baselines
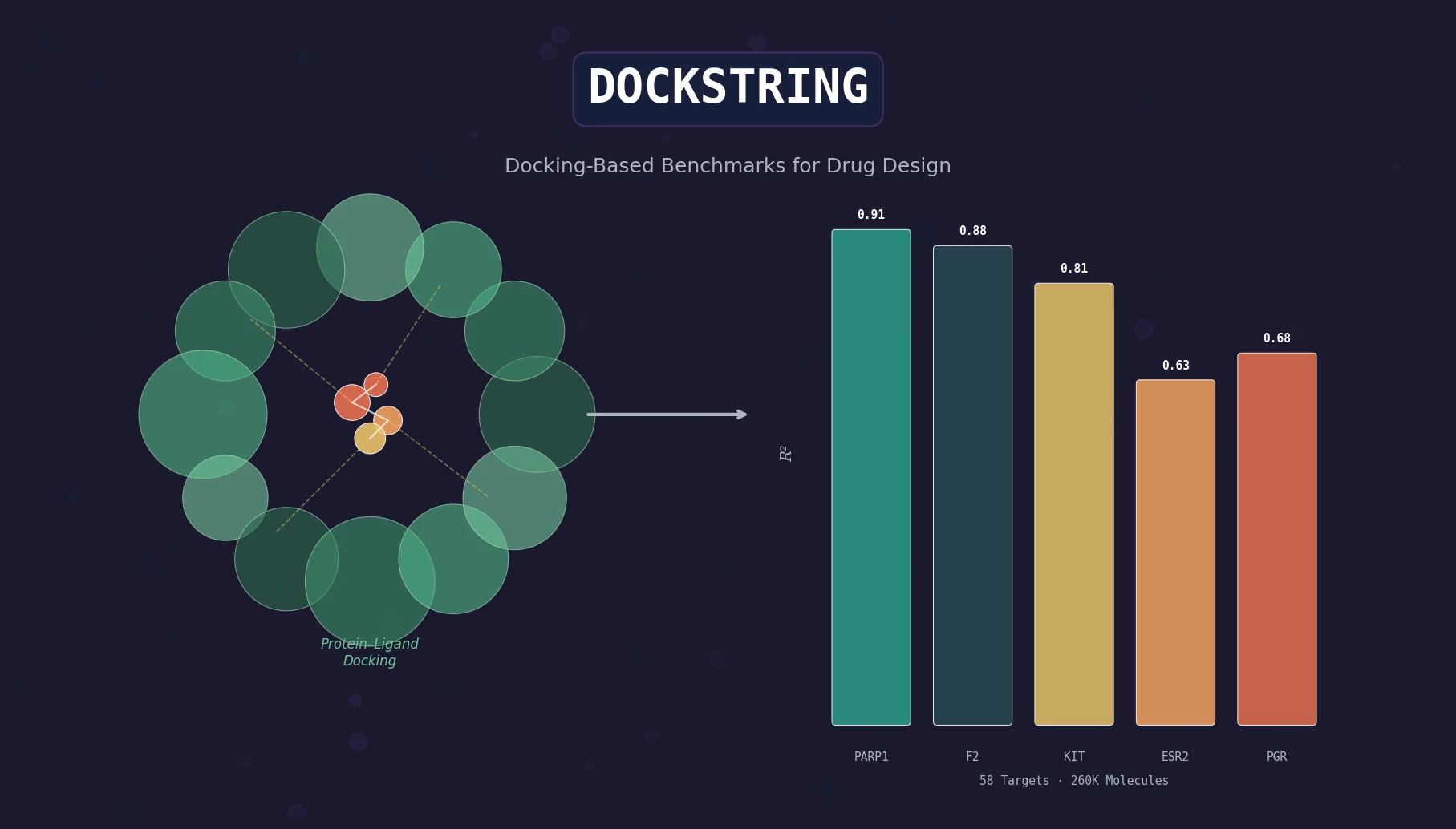
Five targets of varying difficulty were selected: PARP1 (easy), F2 (easy-medium), KIT (medium), ESR2 (hard), and PGR (hard). Baselines include Ridge, Lasso, XGBoost, exact GP, sparse GP, MPNN, and Attentive FP.
| Target | Ridge | Lasso | XGBoost | GP (exact) | GP (sparse) | MPNN | Attentive FP |
|---|---|---|---|---|---|---|---|
| logP | 0.640 | 0.640 | 0.734 | 0.707 | 0.716 | 0.953 | 1.000 |
| QED | 0.519 | 0.483 | 0.660 | 0.640 | 0.598 | 0.901 | 0.981 |
| ESR2 | 0.421 | 0.416 | 0.497 | 0.441 | 0.508 | 0.506 | 0.627 |
| F2 | 0.672 | 0.663 | 0.688 | 0.705 | 0.744 | 0.798 | 0.880 |
| KIT | 0.604 | 0.594 | 0.674 | 0.637 | 0.684 | 0.755 | 0.806 |
| PARP1 | 0.706 | 0.700 | 0.723 | 0.743 | 0.772 | 0.815 | 0.910 |
| PGR | 0.242 | 0.245 | 0.345 | 0.291 | 0.387 | 0.324 | 0.678 |
Values are mean $R^2$ over three runs. Attentive FP achieves the best performance on every target but remains well below perfect prediction on the harder targets, confirming that docking score regression is a meaningful benchmark.
Virtual Screening Baselines
Models trained on PARP1, KIT, and PGR docking scores rank all molecules in ZINC20 (~1 billion compounds). The top 5,000 predictions are docked, and the enrichment factor (EF) is computed relative to a 0.1 percentile activity threshold.
| Target | Threshold | FSS | Ridge | Attentive FP |
|---|---|---|---|---|
| KIT | -10.7 | 239.2 | 451.6 | 766.5 |
| PARP1 | -12.1 | 313.1 | 325.9 | 472.2 |
| PGR | -10.1 | 161.4 | 120.5 | 461.3 |
The maximum possible EF is 1,000. Attentive FP substantially outperforms fingerprint similarity search (FSS) and Ridge regression across all targets.
De Novo Design Baselines
Four optimization methods were tested: SELFIES GA, Graph GA, GP-BO with UCB acquisition ($\beta = 10$), and GP-BO with expected improvement (EI), each with a budget of 5,000 objective function evaluations. Without QED penalties, all methods easily surpass the best training set molecules but produce large, lipophilic, undrug-like compounds. With QED penalties, the tasks become substantially harder: GP-BO with EI is the only method that finds 25 molecules better than the training set across all three tasks.
The Selective JAK2 task proved hardest due to the high correlation between JAK2 and LCK scores. Pose analysis of the top de novo molecule revealed a dual binding mode: type V inhibitor behavior in JAK2 (binding distant N- and C-terminal lobe regions) and type I behavior in LCK (hinge-binding), suggesting a plausible selectivity mechanism.
Key Findings and Limitations
Key findings:
- Docking scores are substantially harder to predict than logP or QED, making them more suitable for benchmarking high-performing ML models. Graph neural networks (Attentive FP) achieve near-perfect $R^2$ on logP but only 0.63-0.91 on docking targets.
- In-distribution regression difficulty does not necessarily predict out-of-distribution virtual screening difficulty. PARP1 is easiest for regression, but KIT is easiest for virtual screening.
- Adding a QED penalty to de novo design objectives transforms trivially solvable tasks into meaningful benchmarks. The adversarial Selective JAK2 objective, which exploits correlated docking scores, may be an effective way to avoid docking score biases toward large and lipophilic molecules.
- Docking scores from related protein targets are highly correlated, supporting the biological meaningfulness of the dataset and enabling multiobjective and transfer learning tasks.
Limitations acknowledged by the authors:
- Docking scores are approximate heuristics. They use static binding sites and force fields with limited calibration for certain metal ions. DOCKSTRING benchmarks should not substitute for rational drug design and experimental validation.
- The pipeline relies on AutoDock Vina specifically; other docking programs may produce different rankings.
- Top de novo molecules for F2 and Promiscuous PPAR contain conjugated ring structures uncommon in successful drugs.
- Platform support is primarily Linux, with noted scoring inconsistencies on macOS.
Future directions mentioned include multiobjective tasks (transfer learning, few-shot learning), improved objective functions for better pharmacokinetic properties and synthetic feasibility, and multifidelity optimization tasks combining docking with more expensive computational methods.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Ligand source | ExCAPE-DB (PubChem + ChEMBL) | 260,155 molecules | Actives against 58 targets + 150K inactive-only |
| Docking scores | DOCKSTRING dataset | 15M+ scores and poses | Full matrix across all molecule-target pairs |
| Virtual screening library | ZINC20 | ~1 billion molecules | Used for out-of-distribution evaluation |
| Target structures | DUD-E + PDB 6CM4 (DRD2) | 58 targets | Kinases (22), enzymes (12), nuclear receptors (9), proteases (7), GPCRs (5), cytochromes (2), chaperone (1) |
Algorithms
- Docking engine: AutoDock Vina with default exhaustiveness (8), up to 9 binding modes, energy range of 3 kcal/mol
- Ligand preparation: Open Babel (protonation at pH 7.4), RDKit ETKDG (3D embedding), MMFF94 (force field refinement), Gasteiger charges
- Regression models: Ridge, Lasso, XGBoost (hyperparameters via 20-configuration random search with 5-fold CV), exact GP and sparse GP (Tanimoto kernel on fingerprints), MPNN, Attentive FP (DeepChem defaults, 10 epochs)
- Optimization: Graph GA (population 250, offspring 25, mutation rate 0.01), SELFIES GA (same population/offspring settings), GP-BO with UCB ($\beta = 10$) or EI (batch size 5, 1000 offspring, 25 generations per iteration)
Evaluation
| Metric | Setting | Notes |
|---|---|---|
| $R^2$ (coefficient of determination) | Regression | Cluster-split train/test |
| EF (enrichment factor) | Virtual screening | Top 5,000 from ZINC20, 0.1 percentile threshold |
| Objective value trajectory | De novo design | 5,000 function evaluation budget |
Hardware
The dataset required over 500,000 CPU hours to compute, using the University of Cambridge Research Computing Service (EPSRC and DiRAC funded). Per-target docking takes approximately 15 seconds on 8 CPUs.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| DOCKSTRING Python package | Code | Apache 2.0 | Wraps AutoDock Vina; available via conda-forge and PyPI |
| DOCKSTRING dataset | Dataset | Apache 2.0 | 15M+ docking scores and poses for 260K molecules x 58 targets |
| Benchmark baselines | Code | Apache 2.0 | Regression, virtual screening, and de novo design baseline implementations |
Paper Information
Citation: García-Ortegón, M., Simm, G. N. C., Tripp, A. J., Hernández-Lobato, J. M., Bender, A., & Bacallado, S. (2022). DOCKSTRING: Easy Molecular Docking Yields Better Benchmarks for Ligand Design. Journal of Chemical Information and Modeling, 62(15), 3486-3502. https://doi.org/10.1021/acs.jcim.1c01334
Publication: Journal of Chemical Information and Modeling, 2022
Additional Resources:
Citation
@article{garciaortegon2022dockstring,
title={{DOCKSTRING}: Easy Molecular Docking Yields Better Benchmarks for Ligand Design},
author={Garc{\'\i}a-Orteg{\'o}n, Miguel and Simm, Gregor N. C. and Tripp, Austin J. and Hern{\'a}ndez-Lobato, Jos{\'e} Miguel and Bender, Andreas and Bacallado, Sergio},
journal={Journal of Chemical Information and Modeling},
volume={62},
number={15},
pages={3486--3502},
year={2022},
publisher={American Chemical Society},
doi={10.1021/acs.jcim.1c01334}
}