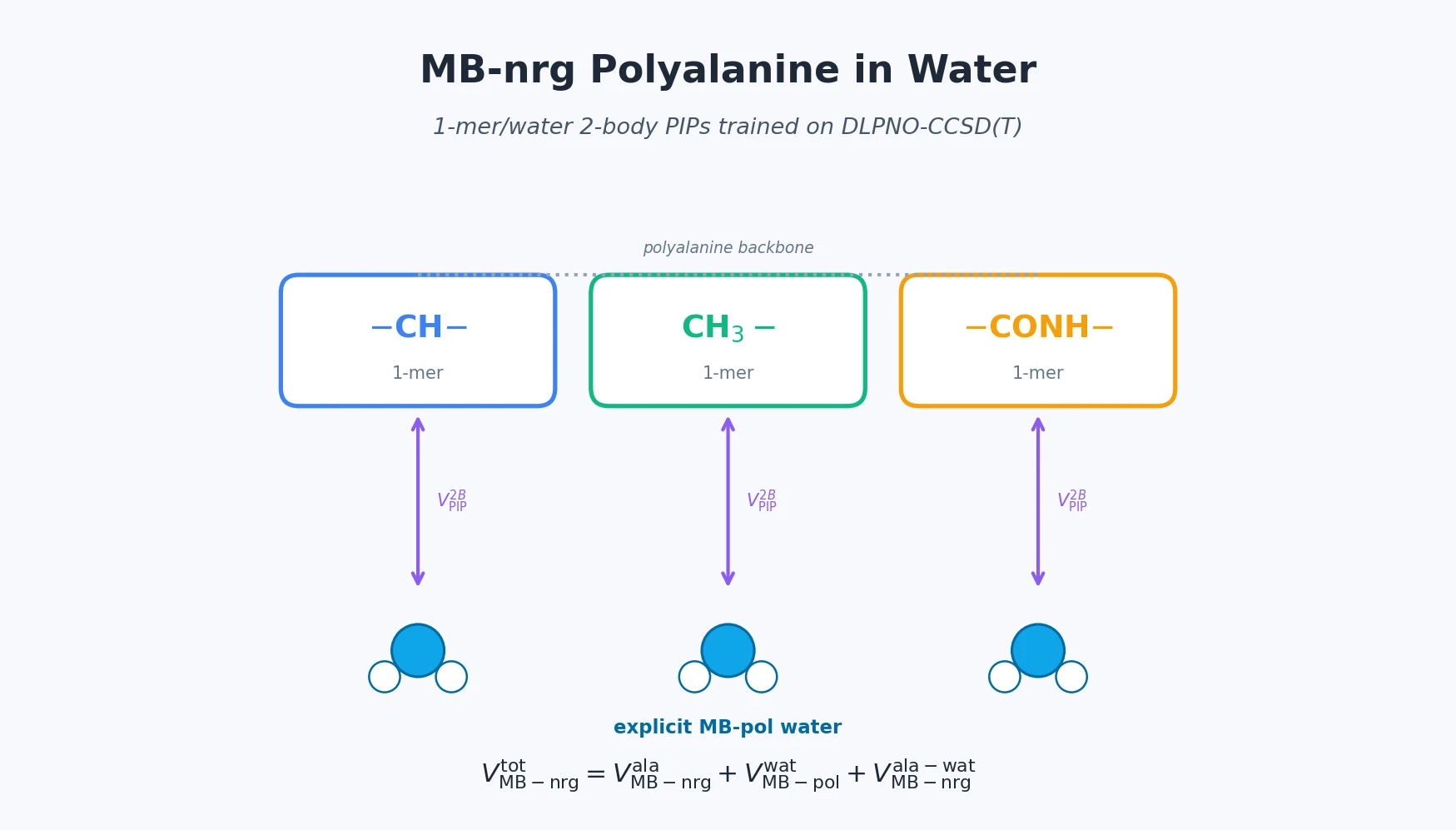
Extending MB-nrg from Gas-Phase Polyalanine to Aqueous Solution
This is a Method paper, the second installment in Zhou and Paesani’s MB-nrg-for-biomolecules series. Paper I (covered in MB-nrg: CCSD(T)-Accurate Potentials for Polyalanine) decomposed gas-phase polyalanine into functional-group $n$-mers and fit permutationally invariant polynomials (PIPs) to DLPNO-CCSD(T)/aug-cc-pVTZ reference data. This sequel adds the missing piece: explicit, machine-learned 2-body interactions between every polyalanine functional-group 1-mer and a water molecule, trained on the same coupled-cluster reference. The resulting PEF couples the gas-phase intramolecular MB-nrg term, the MB-pol water model, and a new MB-nrg ala-water cross term within a single modular many-body decomposition.
Why Empirical Force Fields Struggle with Hydrated Peptides
Biomolecular function in water emerges from a coupling of intramolecular flexibility with solvent-mediated interactions, including hydrogen-bond networks, cooperative polarization, dispersion, and short-range exchange-repulsion. Empirical force fields such as AMBER, CHARMM, and OPLS approximate the multidimensional PES with pairwise-additive analytical terms whose parameters are tuned to experimental observables or low-level quantum data. The authors note that this functional form leads to systematic errors in predicted conformational ensembles for short peptides and intrinsically disordered proteins (IDPs), with reported overpopulation of polyproline II (pPII) basins and antiparallel $\beta$ regions for alanine residues, plus underrepresentation of the transitional $\beta$ basin compared to experiment.
Polarizable force fields recover dielectric and hydration trends through induced dipoles, but still lean on empirical functional forms and miss short-range quantum effects (charge transfer, charge penetration, exchange-repulsion) that arise from electron-density overlap. Machine-learned force fields like MACE-OFF, GEMS, and FeNNix-Bio1 have improved bio-organic accuracy, but they still depend critically on the diversity and quality of training data, struggle to decompose energies into physically interpretable components, and most rely on DFT references that inherit delocalization errors and incomplete long-range correlation. Local descriptors common to MLFFs also limit treatment of long-range electrostatics and many-body correlations, both essential for biomolecular solvation.
The MB-nrg formalism, originally developed for water and small molecules and recently extended to alkanes and gas-phase polyalanine, offers an alternative: a rigorous many-body expansion (MBE) of the energy combined with both data-driven $n$-body PIPs and physics-based long-range terms. Paper II asks whether this modular gas-phase scaffold can be cleanly extended to aqueous environments by adding only short-range peptide-water 2-body PIPs.
A Modular MB-nrg PEF for Polyalanine in Water
The MBE writes the total energy of a system of $N$ 1-mers as
$$ E_N(1, \dots, N) = \sum_{i=1}^{N} \varepsilon^{1\mathrm{B}}(i) + \sum_{i<j}^{N} \varepsilon^{2\mathrm{B}}(i,j) + \sum_{i<j<k}^{N} \varepsilon^{3\mathrm{B}}(i,j,k) + \dots + \varepsilon^{N\mathrm{B}}(1, \dots, N) $$
with each $n$-body term defined recursively as the $n$-mer energy minus all lower-order contributions. The MBE converges quickly for insulating molecular systems with large electronic band gaps (such as water and peptides), so explicit PIP corrections are typically truncated at $n \leq 4$, with higher-order effects absorbed into many-body polarization.
For polyalanine in water, the total potential is partitioned into three modular blocks:
$$ V_{\mathrm{MB\text{-}nrg}}^{\mathrm{tot}} = V_{\mathrm{MB\text{-}nrg}}^{\mathrm{ala}} + V_{\mathrm{MB\text{-}pol}}^{\mathrm{wat}} + V_{\mathrm{MB\text{-}nrg}}^{\mathrm{ala\text{-}wat}} $$
where $V_{\mathrm{MB\text{-}nrg}}^{\mathrm{ala}}$ is the gas-phase intramolecular polyalanine PEF from Paper I, $V_{\mathrm{MB\text{-}pol}}^{\mathrm{wat}}$ is the MB-pol water model, and $V_{\mathrm{MB\text{-}nrg}}^{\mathrm{ala\text{-}wat}}$ is the new peptide-water cross term. The cross term itself follows the MB-nrg recipe of splitting machine-learned and physics-based contributions:
$$ V_{\mathrm{MB\text{-}nrg}}^{\mathrm{ala\text{-}wat}} = V_{\mathrm{ML}} + V_{\mathrm{phys}} $$
with $V_{\mathrm{ML}} = V_{\mathrm{ML}}^{2\mathrm{B}}$ (only 2-body PIPs in this implementation) and $V_{\mathrm{phys}} = V_{\mathrm{elec}} + V_{\mathrm{disp}}$. The 2-body machine-learned term sums switched PIPs over every (1-mer, water) dimer:
$$ V_{\mathrm{ML}}^{2\mathrm{B}} = \sum_{i=1}^{N} s^{2\mathrm{B}}(\mathrm{M}_i, \mathrm{WAT}), V_{\mathrm{PIP}}^{2\mathrm{B}}(\mathrm{M}_i, \mathrm{WAT}) $$
where $\mathrm{M}_i$ is the $i$-th polyalanine functional-group 1-mer (-CH-, CH$_3$-, or -CONH-), WAT is a water molecule, and $s^{2\mathrm{B}}$ is a cosine switching function
$$ s^{2\mathrm{B}}(x) = \begin{cases} 1 & x < 0 \\ (1 + \cos(x))/2 & 0 \leq x < 1 \\ 0 & 1 \leq x \end{cases}, \quad x = \frac{R - R_{\mathrm{in}}}{R_{\mathrm{out}} - R_{\mathrm{in}}} $$
that smoothly attenuates the short-range PIP beyond a defined distance to preserve energy conservation in MD. The physics-based block uses a Thole-modified self-consistent polarization model (inherited from MB-pol) for $V_{\mathrm{elec}}$ and a Tang-Toennies damped dispersion sum
$$ V_{\mathrm{disp}} = -\sum_{\substack{\alpha \in 1\text{-mers} \\ \beta \in \mathrm{water}}} f(\mathrm{b}_{\alpha\beta} R_{\alpha\beta}), \frac{C_{6, \alpha\beta}}{R_{\alpha\beta}^{6}} $$
with $C_{6, \alpha\beta}$ coefficients and atomic polarizabilities derived from the exchange-hole dipole moment (XDM) method, and atomic charges fit to reproduce the permanent multipole moments of each $n$-mer’s optimized structure.
The authors stress that explicit 3-body and higher peptide-water PIPs are deliberately omitted in this first implementation; their effects are absorbed into the classical polarization term. They flag that strongly hydrogen-bonded or cooperative configurations may benefit from adding higher-body corrections in future work, following the precedent of MB-pol(2023) for water.
Training Set Generation and DLPNO-CCSD(T) Reference Data
Training pools for the three 1-mer-water dimers (CH$_3$-H$_2$O, -CH–H$_2$O, -CONH–H$_2$O) extend the parallel-bias metadynamics with partitioned families (PBMetaD+PFs) protocol from Paper I. Covalent boundaries are capped with “ghost” hydrogens at fixed C-H (1.14 Å) and N-H (1.09 Å) distances to preserve closed-shell character; each 2-body energy is referenced to the corresponding optimized capped 1-mer-water geometry to remove constant offsets.
PBMetaD simulations are run in LAMMPS interfaced with PLUMED, using Amber ff14SB for the alanine 1-mers and TIP4P/2005f for water. Collective variables span all heavy-atom bonds, angles, and dihedrals in each dimer. To target distinct interaction regimes, three separate biased runs apply upper and lower walls on the 1-mer/water center-of-mass distance: 0-4 Å (short-range repulsion), 4-7 Å (mid-range attraction), and 7-10 Å (long-range orientation-dependent interactions). Each dimer yields about 600,000 configurations, reduced to roughly 40,000 training and 2,000 test configurations per type by K-means clustering.
Reference 2-body energies are computed at the DLPNO-CCSD(T)/aug-cc-pVTZ level in ORCA, using the aug-cc-pVTZ/C auxiliary basis, the RIJCOSX approximation, TightSCF, TightPNO, and the PModel pair-selection option. The counterpoise method corrects every 2-body energy for basis set superposition error.
Each PIP minimizes a weighted, ridge-regularized least-squares objective:
$$ \chi^2 = \sum_{k \in \mathcal{S}} w_k \left[ V^{2\mathrm{B}}(k) - \varepsilon^{2\mathrm{B}}(k) \right]^2 + \Gamma^2 \sum_l c_l^2 $$
with $\Gamma = 0.0005$ throughout. Training weights bias the fit toward low-energy configurations,
$$ w_k = \left( \frac{\delta E}{\varepsilon^{2\mathrm{B}}(k) - \varepsilon_{\mathrm{min}}^{2\mathrm{B}} + \delta E} \right)^2 $$
with $\delta E = 40$ kcal/mol for all 1-mer-water pairs. MB-Fit handles the optimization, combining simplex minimization for non-linear parameters (Morse decay constants) with ridge regression for the linear coefficients.
Table 1 reports the PIP specifications. All three PIPs use polynomial degree 3 with a complete, unscreened basis. The -CH- and CH$_3$- dimers each require 710 symmetrized terms; the chemically richer -CONH- dimer requires 1,267 terms to capture its dipolar character and directional hydrogen bonding. Training-set sizes range from 41,781 to 43,174 configurations.
| 1-mer type | PIP degree | PIP terms | Training configs | Train RMSD (kcal/mol) | Test RMSD (kcal/mol) | Train MAE | Test MAE |
|---|---|---|---|---|---|---|---|
| -CH- | 3 | 710 | 43,174 | 0.07 | 0.08 | 0.06 | 0.06 |
| CH$_3$- | 3 | 710 | 43,172 | 0.08 | 0.08 | 0.05 | 0.05 |
| -CONH- | 3 | 1,267 | 41,781 | 0.18 | 0.20 | 0.13 | 0.16 |
All RMSDs sit below 0.20 kcal/mol on both train and test splits, well within sub-chemical accuracy.
Validation: Dimer Scans, Free-Energy Surfaces, and Hydration
The authors stage four validation studies of increasing complexity, each touching a distinct facet of the new PEF.
Alanine dipeptide-water dimer scans. One-dimensional scans probe the interaction energy along four hydrogen-bonding coordinates of an alanine dipeptide-water dimer: O$_1$-H$_w$, H$_1$-O$_w$, O$_2$-H$_w$, and H$_2$-O$_w$, where subscripts 1 and 2 mark the acetyl and N-methyl termini. The dipeptide is constrained to four representative Ramachandran conformations: C5 ($\varphi = -150°$, $\psi = 150°$), pPII ($\varphi = -80°$, $\psi = 150°$), C7$_{\mathrm{eq}}$ ($\varphi = -80°$, $\psi = 70°$), and right-handed $\alpha$-helix $\alpha_R$ ($\varphi = -80°$, $\psi = -30°$). MB-nrg closely tracks the DLPNO-CCSD(T)/aug-cc-pVTZ reference curves across all 16 (4 conformation $\times$ 4 site) scans, despite never seeing the full dipeptide-water surface during training. Amber ff14SB/TIP3P and ff19SB/OPC underestimate hydrogen-bond depths and miss curvature near equilibrium, with the ff14SB/TIP3P combination yielding slightly better overall agreement than ff19SB/OPC even though TIP3P is the less accurate water model.
Two specific failure modes of the empirical force fields stand out. In the pPII conformation, both ff14SB and ff19SB predict significantly deeper interaction wells than the reference, overstabilizing several hydrogen bonds. In the H$_2$-O$_w$ scan of the $\alpha_R$ conformation, both empirical FFs exhibit a spurious 2.5-4.0 Å energy barrier that the authors trace to the simple Lennard-Jones repulsion between the acetyl carbonyl oxygen and water; MB-nrg and DLPNO-CCSD(T) instead show a smoothly decaying profile. The one MB-nrg deviation noted is the C5 H$_1$-O$_w$ scan in the 1.5-2.5 Å range, where MB-nrg predicts a slightly more attractive interaction than the reference. Here the H$_1$-O$_2$ distance is 2.3 Å and water acts simultaneously as acceptor at H$_1$ and donor to O$_2$, a cooperative pattern the authors expect would require explicit 2-mer-water or 3-mer-water terms to fully reproduce.
Free-energy surface in explicit MB-pol water. Four-walker well-tempered metadynamics (WT-MetaD) simulations explore the conformational landscape of alanine dipeptide as a function of $(\varphi, \psi)$, biasing the central alanine residue’s backbone dihedrals every 500 steps with 1.0 kJ/mol Gaussians of 11.46° width. The free-energy section reports 2.5 ns per replica across four parallel walkers (10 ns aggregate, matching the Figure 6 caption); the methods section states 8 ns total, an internal inconsistency in the paper. The MB-nrg FES recovers all major low-energy conformers identified by NMR and prior MP2/DFT studies: a global minimum at $\alpha_R$, additional local minima in C5, $\beta_2$, and $\alpha_L$, and a metastable pPII basin. The C7$_{\mathrm{eq}}$ minimum that dominates the gas-phase Ramachandran surface in Paper I is significantly destabilized in solution, consistent with experiment.
Quantitatively, MB-nrg predicts $\alpha_R$ and $\beta_2$ as isoenergetic global minima, with C5 about 3 kcal/mol higher in free energy. Prior DFT-with-implicit-solvation studies (Mironov et al., Yang and Honig) report C5, $\alpha_R$, and $\beta_2$ as nearly isoenergetic, and the authors note that the discrepancy may reflect the explicit MB-pol water treatment, residual DFT errors in the reference, or both. They flag a planned systematic benchmarking of MB-nrg PEFs for diverse polypeptides against both DFT and DLPNO-CCSD(T) data in future work. The Amber FESs over-stabilize pPII relative to C5/$\alpha_R$, contradicting experimental and DFT benchmarks; ff19SB/OPC also exhibits a spurious C7$_{\mathrm{eq}}$ minimum that is absent from MB-nrg.
Hydration radial distribution functions. Site-site RDFs at 300 K for the same hydrogen-bond contacts (O$_1$-H$_w$, O$_2$-H$_w$, H$_1$-O$_w$, H$_2$-O$_w$) are computed from NVT MD trajectories. All three models reproduce well-defined first-shell peaks near 2.0 Å. For the O-H$_w$ pairs, MB-nrg shows a broader, slightly right-shifted second-shell peak, indicating less rigid water structure beyond the first shell. The amide-hydrogen RDFs are nearly identical between ff14SB/TIP3P and ff19SB/OPC, while MB-nrg reveals subtle first-shell shifts (shorter H$_1$-O$_w$, longer H$_2$-O$_w$) and weaker, less-defined second-shell features near 3.7-3.8 Å that are absent from the empirical force fields and consistent with prior ab initio MD on alanine dipeptide.
A Modular Path to Chemically Accurate Biomolecular Simulations
Across the four benchmarks, the same picture emerges: a modular, bottom-up MB-nrg PEF built from functional-group $n$-mers and trained only on isolated 1-mer-water dimers can reach DLPNO-CCSD(T) accuracy for both energetic and structural observables of alanine dipeptide in explicit water. The decomposition into a gas-phase intramolecular term, an MB-pol water model, and an MB-nrg cross term keeps each piece interpretable and individually replaceable; the gas-phase polyalanine PEF from Paper I drops in unchanged, and the new ala-water PIPs were fit without ever seeing the full alanine dipeptide-water PES.
The authors are explicit about limitations:
- The cross term currently includes only 2-body PIPs (one 1-mer with one water). Higher-body peptide-water terms ($n > 2$) are folded into the classical polarization, which the authors expect will be inadequate for strongly cooperative configurations such as the C5 H$_1$-O$_w$ scan where one water bridges H$_1$ and O$_2$.
- Quantitative differences between the MB-nrg FES and prior implicit-solvation DFT studies (relative depths of $\alpha_R$, $\beta_2$, and C5) remain to be reconciled through systematic benchmarking against higher-level reference data.
- Only polyalanine is considered. The framework is designed to generalize to other amino acids and side-chain-water interactions, but sequence- and side-chain-specific PIPs are still to be fit.
- No public release of the parameterized PEF or training data is announced; the data availability statement says “available from the authors upon request.”
The paper positions MB-nrg as a transferable, interpretable strategy for chemically accurate biomolecular simulations in solution, with future work aimed at heteropolypeptides and explicit higher-order many-body cross terms.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training pools | PBMetaD+PFs in LAMMPS/PLUMED | ~600,000 configs per dimer, reduced to ~40,000 | ff14SB for alanine 1-mers, TIP4P/2005f for water; 300 K, 500 K, 700 K |
| Distance regimes | Walls on 1-mer/water COM distance | 0-4, 4-7, and 7-10 Å | Short-range repulsion, mid-range attraction, long-range orientation |
| Training labels | DLPNO-CCSD(T)/aug-cc-pVTZ in ORCA | 3 unique 1-mer-water dimer types | RIJCOSX, TightSCF, TightPNO, PModel; counterpoise BSSE correction |
| Test sets | Held-out clustered configs | ~2,000 per dimer | Same K-means clustering protocol |
| Alanine dipeptide-water scans | 1D scans along 4 H-bond coordinates in 4 conformations | 16 scans total | C5, pPII, C7$_{\mathrm{eq}}$, and $\alpha_R$ conformations |
| Alanine dipeptide FES | WT-MetaD on $\varphi$, $\psi$ in MB-pol water | 4 walkers, 2.5 ns each (10 ns total per the results section and Figure 6 caption; methods section states 8 ns) | 1.0 kJ/mol height, 11.46° width, deposition every 500 steps |
| Hydration RDFs | NVT MD at 300 K | Single trajectory per model | Same H-bond sites as the dimer scans |
Per the data availability statement, “any data generated and analyzed in this study, including the MB-nrg PEF, are available from the authors upon request.” The MBX engine is publicly available on GitHub under a UC Regents custom license that grants free use for educational, research, and non-profit purposes but restricts commercial use. No public release of the new ala-water PIPs is announced in the text.
Artifacts table
| Artifact | Type | License | Notes |
|---|---|---|---|
| MBX | Code | UC Regents custom (academic/non-profit only; no SPDX-recognized OSS license) | C++ many-body potential engine; runs the MB-nrg PEF via LAMMPS and PLUMED |
| MB-Fit | Code | Check repo | Training pipeline for PIP fitting; used to fit the new 1-mer-water PIPs |
| MB-nrg ala-water PIPs (this paper) | Model | Not released | “Available from the authors upon request” per the data availability statement |
| DLPNO-CCSD(T) training/test sets | Dataset | Not released | Same statement; ~600,000 raw configs per dimer reduced to ~40,000 train + ~2,000 test |
Algorithms
- Many-body expansion of the energy partitioned into three modular blocks: $V_{\mathrm{MB\text{-}nrg}}^{\mathrm{ala}} + V_{\mathrm{MB\text{-}pol}}^{\mathrm{wat}} + V_{\mathrm{MB\text{-}nrg}}^{\mathrm{ala\text{-}wat}}$.
- Cross term split into $V_{\mathrm{ML}}^{2\mathrm{B}}$ (PIPs over every 1-mer-water dimer) and $V_{\mathrm{phys}} = V_{\mathrm{elec}} + V_{\mathrm{disp}}$.
- Permutationally invariant polynomials in Morse-exponential variables $\xi_{ij} = \exp(-k_{\tau(ij)} R_{ij})$, symmetrized over chemically equivalent atoms; same construction as the NMA-water PIPs.
- Cosine switching function $s^{2\mathrm{B}}$ smoothly attenuates short-range PIPs between user-defined inner and outer cutoffs.
- Dispersion: Tang-Toennies damped $C_6/R^6$ with XDM-derived coefficients and damping parameters.
- Electrostatics: modified Thole model with self-consistent induced dipoles for many-body polarization; per-atom charges fit to reproduce permanent multipole moments of each $n$-mer’s optimized structure.
- Ghost-H capping at cleaved covalent boundaries with fixed C-H (1.14 Å) and N-H (1.09 Å) distances; per-dimer optimized-structure referencing.
- Training with simplex minimization for non-linear parameters and ridge regression for linear coefficients via MB-Fit, with low-energy weighting and $\Gamma = 0.0005$, $\delta E = 40$ kcal/mol.
- WT-MetaD with four parallel walkers for the alanine dipeptide FES.
Models
- Three new 1-mer-water 2-body PIPs covering -CH-/H$_2$O, CH$_3$-/H$_2$O, and -CONH-/H$_2$O dimers.
- All three PIPs use polynomial degree 3 with a complete, unscreened basis (no term screening).
- Term counts: 710 for -CH-/H$_2$O and CH$_3$-/H$_2$O, 1,267 for -CONH-/H$_2$O.
- Combined with the gas-phase polyalanine MB-nrg PEF from Paper I and the MB-pol water model, exercised through MBX, LAMMPS, and PLUMED.
Evaluation
| Metric | MB-nrg | Amber ff14SB/TIP3P | Amber ff19SB/OPC |
|---|---|---|---|
| -CH-/H$_2$O 2-body train/test RMSD | 0.07 / 0.08 kcal/mol | n/a | n/a |
| CH$_3$-/H$_2$O 2-body train/test RMSD | 0.08 / 0.08 kcal/mol | n/a | n/a |
| -CONH-/H$_2$O 2-body train/test RMSD | 0.18 / 0.20 kcal/mol | n/a | n/a |
| Alanine dipeptide-water 1D scans (qualitative) | Tracks DLPNO-CCSD(T) curves across 16 scans | Underestimates H-bond depths; spurious $\alpha_R$ H$_2$-O$_w$ barrier | Same shape as ff14SB/TIP3P |
| Alanine dipeptide FES global minima | Isoenergetic $\alpha_R$ and $\beta_2$; C5 ~3 kcal/mol higher | Over-stabilizes pPII | Over-stabilizes pPII; spurious C7$_{\mathrm{eq}}$ minimum |
| O-H$_w$ second shell | Broader, right-shifted; finer detail consistent with prior AIMD | Sharper, less detail | Sharper, less detail |
| H-O$_w$ second shell | Weak features near 3.7-3.8 Å | Absent | Absent |
Quantitative RMSD or KL-divergence values for the FES and RDF benchmarks are not reported in the main text.
Hardware
The authors acknowledge support from the Air Force Office of Scientific Research (FA9550-20-1-0351, theoretical development) and NSF (award 2311260, MBX implementation). Computational resources came from the DoD High Performance Computing Modernization Program, the San Diego Supercomputer Center via ACCESS allocation CHE240114, and NERSC (contract DE-AC02-05CH11231, award BES-ERCAP0030920). Specific wall-clock and node-hour figures are not reported in the main text.
Paper Information
Citation: Zhou, R., & Paesani, F. (2025). Toward Chemical Accuracy in Biomolecular Simulations through Data-Driven Many-Body Potentials: II. Polyalanine in Water. ChemRxiv. https://doi.org/10.26434/chemrxiv-2025-j6cwv-v2
Publication: ChemRxiv preprint (version 2), 10 October 2025.
Additional Resources:
- MBX software (Paesani group)
- MB-Fit (training pipeline)
- Companion paper: MB-nrg: CCSD(T)-Accurate Potentials for Polyalanine (Paper I)
@article{zhou2025toward,
title={Toward Chemical Accuracy in Biomolecular Simulations through Data-Driven Many-Body Potentials: II. Polyalanine in Water},
author={Zhou, Ruihan and Paesani, Francesco},
journal={ChemRxiv},
year={2025},
doi={10.26434/chemrxiv-2025-j6cwv-v2}
}