A Modular MB-nrg Method for Biomolecular Potentials
This is a Method paper. Zhou and colleagues extend the MB-nrg (many-body energy) formalism to covalently bonded biomolecules and build the first coupled-cluster-accurate potential energy function (PEF) for polyalanine in the gas phase. The contribution has three parts: a generalization of the MB-nrg decomposition from whole-molecule 1-mers to functional-group “natural building blocks,” a DLPNO-CCSD(T)/aug-cc-pVTZ training protocol driven by parallel-bias metadynamics sampling, and a demonstration that the resulting PEF reproduces alanine dipeptide energetics and AceAla$_9$Nme secondary-structure dynamics more faithfully than the Amber ff14SB and ff19SB force fields.
Why Empirical Force Fields Fall Short for Protein Dynamics
Protein dynamics span femtosecond vibrations to millisecond conformational changes, and capturing them at atomic resolution is central to understanding catalysis, allostery, and ligand binding. Classical force fields such as CHARMM, OPLS, and Amber approximate the potential energy surface with pairwise-additive analytical terms. This functional form struggles with the many-body interactions that shape disordered regions of proteins, including exchange-repulsion, charge transfer, charge penetration, and cooperative hydrogen bonding. Polarizable force fields add induced dipoles but remain empirically parameterized and fail to capture short-range many-body effects from electron-density overlap.
Quantum-mechanical methods avoid this, but coupled cluster theory scales as $\mathcal{O}(N^7)$ in the number of electrons and even DFT remains $\mathcal{O}(N^3)$ to $\mathcal{O}(N^4)$, ruling out direct ab initio molecular dynamics for biomolecules. Fragmentation methods like molecular fractionation with conjugate caps (MFCC) mitigate the cost, but they truncate the many-body expansion at two bodies and miss long-range hydrogen bonding. Machine-learned force fields (MLFFs) reach near-QM accuracy at lower cost, yet they typically train on DFT data (inheriting delocalization errors and poor dispersion), struggle with interpretability, and extrapolate unreliably. Existing permutationally invariant polynomial (PIP) approaches scale factorially in the number of atoms, capping direct applicability at roughly ten to fifteen atoms per fragment.
MB-nrg PEFs based on the many-body expansion and PIPs have successfully modeled water, halides in water, carbon dioxide, methane, ammonia, nitrogen pentoxide, and N-methylacetamide. Extending them to covalently bonded biomolecules requires rethinking what counts as a “body.”
Building Polyalanine from Functional-Group n-mers
The MB-nrg formalism starts from the many-body expansion of the total energy,
$$ E_N(1, \dots, N) = \sum_{i=1}^{N} \varepsilon^{1\mathrm{B}}(i) + \sum_{i<j}^{N} \varepsilon^{2\mathrm{B}}(i,j) + \sum_{i<j<k}^{N} \varepsilon^{3\mathrm{B}}(i,j,k) + \dots + \varepsilon^{N\mathrm{B}}(1, \dots, N) $$
where each $n$-body contribution is defined recursively as the $n$-mer energy minus all lower-order terms. The full PEF combines physics-based and data-driven components,
$$ V_{\mathrm{MB\text{-}nrg}} = V_{\mathrm{ML}} + V_{\mathrm{phys}} $$
with $V_{\mathrm{ML}} = V_{\mathrm{ML}}^{1\mathrm{B}} + V_{\mathrm{ML}}^{2\mathrm{B}} + V_{\mathrm{ML}}^{3\mathrm{B}}$ capturing short-range quantum-mechanical interactions, and $V_{\mathrm{phys}} = V_{\mathrm{elec}} + V_{\mathrm{disp}} + V_{\mathrm{rep}}$ supplying electrostatics, dispersion, and repulsion. Dispersion follows a Tang-Toennies damped $C_6/R^6$ form with XDM-derived coefficients; electrostatics uses a Thole-modified self-consistent polarization model inherited from MB-pol; the repulsion term is a Lennard-Jones $R^{-12}$ contribution borrowed from Amber ff14SB, activated only for non-bonded atom pairs not covered by a PIP.
Each data-driven $n$-body term is expressed as
$$ V_{\mathrm{ML}}^{n\mathrm{B}} = \sum_{\mathrm{M}_1 < \dots < \mathrm{M}_n}^{N} s^{n\mathrm{B}}(\mathrm{M}_1, \dots, \mathrm{M}_n), V_{\mathrm{PIP}}^{n\mathrm{B}}(\mathrm{M}_1, \dots, \mathrm{M}_n) $$
where $V_{\mathrm{PIP}}^{n\mathrm{B}}$ is a permutationally invariant polynomial in Morse-like variables $\xi_{ij} = \exp(-k_{\tau(ij)} R_{ij})$ and $s^{n\mathrm{B}}$ is a switching function.
The key extension in this paper, building on earlier work on linear alkanes, is to treat functional groups (not whole molecules) as 1-mers. An Ace-capped, Nme-capped polyalanine chain decomposes into three distinct 1-mer types (-CH-, CH$_3$-, -CONH-), five distinct 2-mer types, and six distinct 3-mer types, for 14 unique PIPs that cover every $n$-mer appearing in any AceAla$_n$Nme chain. Cleaving covalent bonds between 1-mers would produce radicals, so the authors cap dangling valences with “ghost” hydrogen atoms at fixed C-H (1.14 Å) and N-H (1.09 Å) distances. Each $n$-mer energy is then referenced to its own optimized H-capped structure,
$$ E_n(1, \dots, n) = E_n^{\mathrm{H\text{-}capped}}(1, \dots, n) - E_n^{\mathrm{H\text{-}capped,opt}}(1, \dots, n). $$
In the current implementation, only covalently bonded $n$-mers receive PIPs, the 2-body contribution from a dimer with one intervening 1-mer is folded into the corresponding 3-body term, and non-bonded 1-mers interact through the Lennard-Jones repulsion alone. Crucially, no whole-chain polyalanine data enters any stage of training: every PIP is parameterized on isolated $n$-mer configurations, and the total energy is reconstructed through the many-body expansion.
Training on DLPNO-CCSD(T) with Metadynamics Sampling
Training sets are generated for each of the 14 $n$-mer types using parallel-bias metadynamics (PBMetaD) with partitioned families, biasing heavy-atom bonds, angles, and dihedrals across 300 K, 500 K, and 700 K in LAMMPS interfaced with PLUMED and modified OPLS/CM1A and Amber ff14SB force fields. For each $n$-mer, 200,000 candidate configurations are sampled, then reduced to roughly 10,000-20,000 training configurations (and about 1,000 test configurations) through Mini-batch K-means clustering on chemically equivalent pairwise distances. Reference energies are computed at the DLPNO-CCSD(T)/aug-cc-pVTZ level in ORCA.
Each PIP minimizes a weighted, ridge-regularized sum of squared errors,
$$ \chi^2 = \sum_{k \in \mathcal{S}} w_k \left[ V^{n\mathrm{B}}(k) - \varepsilon^{n\mathrm{B}}(k) \right]^2 + \Gamma^2 \sum_l c_l^2 $$
with $\Gamma = 0.0005$ throughout and low-energy bias weights
$$ w_k = \left( \frac{\delta E}{\varepsilon^{n\mathrm{B}}(k) - \varepsilon^{n\mathrm{B}}_{\min} + \delta E} \right)^2. $$
MB-Fit handles the fit, combining simplex optimization for non-linear parameters $k_{\tau(ij)}$ with ridge regression for the linear coefficients $c_l$.
Table 1 in the paper reports, for each of the 14 PIPs, the polynomial degree (5 for the smaller -CH- and CH$_3$- 1-mers, 3 for the larger -CONH- 1-mer and for all 2-mers and 3-mers), the number of symmetrized monomials (ranging from 635 for the -CH- and CH$_3$- 1-mers to 2871 for the -CONH-CH-CONH- 3-mer), the training-set size, and RMSDs for the train and test splits. All training RMSDs stay below 0.4 kcal/mol and all test RMSDs below 0.5 kcal/mol, with the smallest errors for the -CH- and CH$_3$- 1-mers (0.05 kcal/mol train, 0.14 kcal/mol test) and the largest test RMSD (0.47 kcal/mol) for the -CONH-CH- 2-mer.
MD validations run in LAMMPS interfaced with MBX and PLUMED. For alanine dipeptide metadynamics, bias potentials on the backbone $\varphi$ and $\psi$ angles are deposited every 500 steps with a 1.0 kJ/mol height and 11.46° width over 10 ns trajectories in the NVT ensemble, using the velocity-Verlet integrator with a 0.5 fs time step. Analogous MetaD runs with Amber ff14SB and ff19SB are performed in Amber23. The longer AceAla$_9$Nme trajectories start from fully extended structures and run in a 100 Å × 100 Å × 100 Å gas-phase box.
CCSD(T) Energy Landscapes, Free-Energy Surfaces, and Helix Dynamics
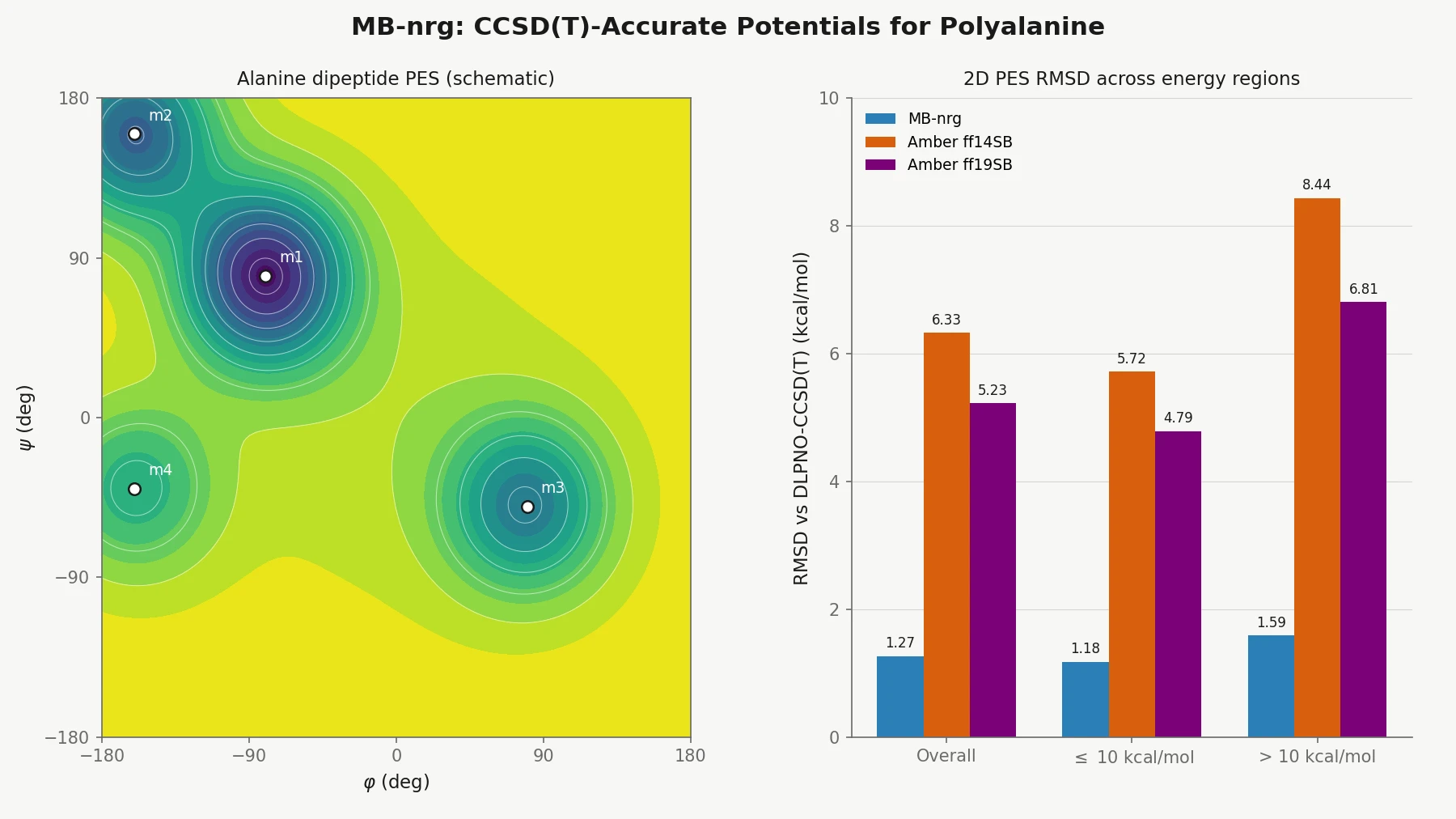
Alanine dipeptide 2D PES. Alanine dipeptide geometries are optimized on a Ramachandran grid with 10° spacing at the RI-MP2/def2-TZVP level and then evaluated at DLPNO-CCSD(T)/aug-cc-pVTZ. Despite never seeing whole alanine dipeptide in training, MB-nrg closely matches the reference locations and relative energies of four minima ($m_1$ to $m_4$), three maxima ($M_1$ to $M_3$), and one saddle point ($X$). Amber ff14SB and ff19SB capture the minima reasonably but badly overshoot the barriers: at $M_1$, MB-nrg misses the reference by only -2.41 kcal/mol, while ff14SB and ff19SB overshoot by +7.50 and +7.83 kcal/mol. The authors also note that ff19SB incorrectly orders the secondary minima by predicting $m_3$ lower than $m_2$.
| Model | RMSD overall (kcal/mol) | RMSD $\leq 10$ kcal/mol | RMSD $> 10$ kcal/mol |
|---|---|---|---|
| MB-nrg | 1.27 | 1.18 | 1.59 |
| Amber ff14SB | 6.33 | 5.72 | 8.44 |
| Amber ff19SB | 5.23 | 4.79 | 6.81 |
The authors attribute MB-nrg’s residual high-energy error to terminal methyl groups approaching the backbone in conformations where non-bonded 1-mer interactions are modeled by the simple LJ repulsion rather than an explicit PIP.
Harmonic vibrations. Normal modes for the $m_1$ and $m_4$ alanine dipeptide conformers, computed by diagonalizing the Hessian, match RI-MP2/def2-TZVP references with mean deviations of 17.41 cm$^{-1}$ and 21.07 cm$^{-1}$ across all 60 modes. The authors acknowledge that some of this discrepancy reflects differences in theoretical levels (MB-nrg is trained to CCSD(T)/aug-cc-pVTZ, while the reference normal modes are computed at RI-MP2/def2-TZVP).
Free-energy surfaces. Well-tempered metadynamics at 300 K produces 2D free-energy surfaces over $(\varphi, \psi)$. MB-nrg yields a smoother FES whose extrema line up with the DLPNO-CCSD(T) reference PES. Amber ff14SB and ff19SB remain reasonable near the low-energy $m_1$ and $m_2$ minima but systematically overestimate the barriers near $M_1$, $M_2$, and $M_3$, which the authors argue artificially confines the dipeptide and suppresses conformational transitions.
Secondary structure in AceAla$_9$Nme. In 600 ps NVT MD starting from a fully extended structure, the STRIDE algorithm tracks residue-level secondary structures. Amber ff14SB and ff19SB collapse into $\alpha$-helices at roughly 40 ps and 80 ps, respectively, with ff19SB remaining especially rigid. MB-nrg takes about 100 ps before helices begin to form and then exhibits continuous oscillations between $3_{10}$- and $\alpha$-helical conformations. Ramachandran plots over the nine alanine residues show MB-nrg exploring the “bridge” region ($\varphi < 0°$, $-20° \leq \psi \leq 20°$) associated with $3_{10}$-helices and sampling the left-handed $\alpha_L$ region that Amber rarely visits. The authors tie this flexibility to experimental observations of alanine-rich peptides in the gas phase and to similar predictions from GEMS and MACE-OFF.
Transferability Without Whole-Chain Training Data
The paper demonstrates that a modular, bottom-up PEF built from functional-group $n$-mers can reach CCSD(T) accuracy for polyalanine in the gas phase without ever training on whole-chain data. Truncating explicit data-driven terms at the 3-body level appears to balance cost and fidelity, with long-range effects handled by many-body polarization in $V_{\mathrm{elec}}$ and by Amber-derived repulsion between distant 1-mers. The 2D PES, harmonic frequencies, free-energy surface, and secondary-structure dynamics each validate a different facet of the model.
The authors are explicit about limitations. The current PEF applies only to gas-phase polyalanine; solvent effects and other amino acids remain open. The Lennard-Jones repulsion for non-bonded 1-mers is a placeholder for eventual 2-body PIPs that should capture short-range interactions during folding. Long-range hydrogen bonding in compact secondary structures (π-helices, $3_{10}$-helices, $\alpha$-helices) may produce non-negligible higher-order many-body contributions that the current 3-body truncation omits. The 2-body contribution from a dimer with one intervening monomer is currently folded into the 3-body term because of steric conflicts between capping hydrogens, and a systematic fix is flagged for future work. The authors position this paper as the first in a series (the “I.” in the title refers to “Polyalanine in the Gas Phase”) that will extend MB-nrg to broader biomolecular systems under physiological conditions. The follow-up, MB-nrg in Solution: Polyalanine in Water with CCSD(T) PEFs, adds explicit 1-mer/water 2-body PIPs and benchmarks alanine dipeptide solvation.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training | Per $n$-mer pools from PBMetaD in LAMMPS/PLUMED | 200,000 configurations each, reduced to ~10-20k via Mini-batch K-means | OPLS/CM1A and Amber ff14SB sampled at 300 K, 500 K, 700 K |
| Training labels | DLPNO-CCSD(T)/aug-cc-pVTZ in ORCA | 14 unique $n$-mer types | Domain-based local pair natural orbital approximation to canonical CCSD(T) |
| Test | Held-out $n$-mer configurations | ~1,000 per $n$-mer | Same clustering protocol |
| Alanine dipeptide benchmark | Ramachandran grid at 10° spacing, RI-MP2/def2-TZVP geometries | 1,296 grid points (approximate) | Single-point energies at DLPNO-CCSD(T)/aug-cc-pVTZ, ff14SB, ff19SB, MB-nrg |
| AceAla$_9$Nme dynamics | 600 ps NVT MD from fully extended start | Single trajectory per model | STRIDE for secondary-structure assignment |
Per the Data Availability statement, “any data generated and analyzed in this study are available from the authors upon request.” No public release is announced in the text.
Algorithms
- Many-body expansion of the energy with 1-, 2-, and 3-body data-driven terms.
- Permutationally invariant polynomials in Morse-exponential variables $\xi_{ij} = \exp(-k_{\tau(ij)} R_{ij})$, symmetrized over chemically equivalent atoms.
- “Ghost” H-capping at cleaved covalent bonds, with fixed C-H (1.14 Å) and N-H (1.09 Å) bond lengths and per-$n$-mer optimized-structure referencing.
- Non-linear parameters fit by simplex minimization, linear coefficients by ridge regression with $\Gamma = 0.0005$.
- Low-energy weighting in the loss through $w_k = (\delta E / (\varepsilon^{n\mathrm{B}}(k) - \varepsilon^{n\mathrm{B}}_{\min} + \delta E))^2$.
- Tang-Toennies damped dispersion with XDM-derived $C_6$ and damping parameters, Thole-modified many-body polarization, and LJ repulsion borrowed from Amber ff14SB.
Models
- 14 PIPs total covering three 1-mer types, five 2-mer types, and six 3-mer types. Polynomial degree is 5 for the -CH- and CH$_3$- 1-mers, and 3 for the -CONH- 1-mer together with all 2-mers and 3-mers. Term counts range from 635 (-CH-, CH$_3$-) to 2871 (-CONH-CH-CONH-).
- MB-nrg PEF implemented in the MBX code and exercised through LAMMPS and PLUMED.
- Training set sizes per $n$-mer range from roughly 12,000 to 47,000 configurations (the -CONH- 1-mer dataset is the largest at 47,438).
Evaluation
| Metric | MB-nrg | Amber ff14SB | Amber ff19SB |
|---|---|---|---|
| $n$-mer training RMSD | $\leq 0.35$ kcal/mol | n/a | n/a |
| $n$-mer test RMSD | $\leq 0.47$ kcal/mol | n/a | n/a |
| Alanine dipeptide 2D PES RMSD (overall) | 1.27 kcal/mol | 6.33 kcal/mol | 5.23 kcal/mol |
| Same, $\leq 10$ kcal/mol region | 1.18 kcal/mol | 5.72 kcal/mol | 4.79 kcal/mol |
| Same, $> 10$ kcal/mol region | 1.59 kcal/mol | 8.44 kcal/mol | 6.81 kcal/mol |
| Alanine dipeptide $m_1$ normal-mode mean deviation vs RI-MP2/def2-TZVP | 17.41 cm$^{-1}$ | n/a | n/a |
| Alanine dipeptide $m_4$ normal-mode mean deviation vs RI-MP2/def2-TZVP | 21.07 cm$^{-1}$ | n/a | n/a |
| AceAla$_9$Nme helix-formation onset (from extended start) | ~100 ps ($\alpha$/$3_{10}$ mix) | ~40 ps ($\alpha$) | ~80 ps ($\alpha$) |
Hardware
Computational resources came from the Air Force Office of Scientific Research (FA9550-20-1-0351), NSF award 2311260, the DoD High Performance Computing Modernization Program, the San Diego Supercomputer Center via ACCESS allocation CHE240114, and NERSC (contract DE-AC02-05CH11231, award BES-ERCAP0030920). Specific wall-clock and node-hour figures are not reported in the main text.
Paper Information
Citation: Zhou, R., Bull-Vulpe, E. F., Pan, Y., & Paesani, F. (2025). Data-Driven Many-Body Simulations of Biomolecules with CCSD(T) Accuracy: I. Polyalanine in the Gas Phase. ChemRxiv. https://doi.org/10.26434/chemrxiv-2025-b05k5
Publication: ChemRxiv preprint, 25 March 2025.
Additional Resources:
@misc{zhou2025data,
title={Data-Driven Many-Body Simulations of Biomolecules with CCSD(T) Accuracy: I. Polyalanine in the Gas Phase},
author={Zhou, Ruihan and Bull-Vulpe, Ethan F. and Pan, Yuanhui and Paesani, Francesco},
year={2025},
doi={10.26434/chemrxiv-2025-b05k5},
howpublished={\url{https://doi.org/10.26434/chemrxiv-2025-b05k5}}
}