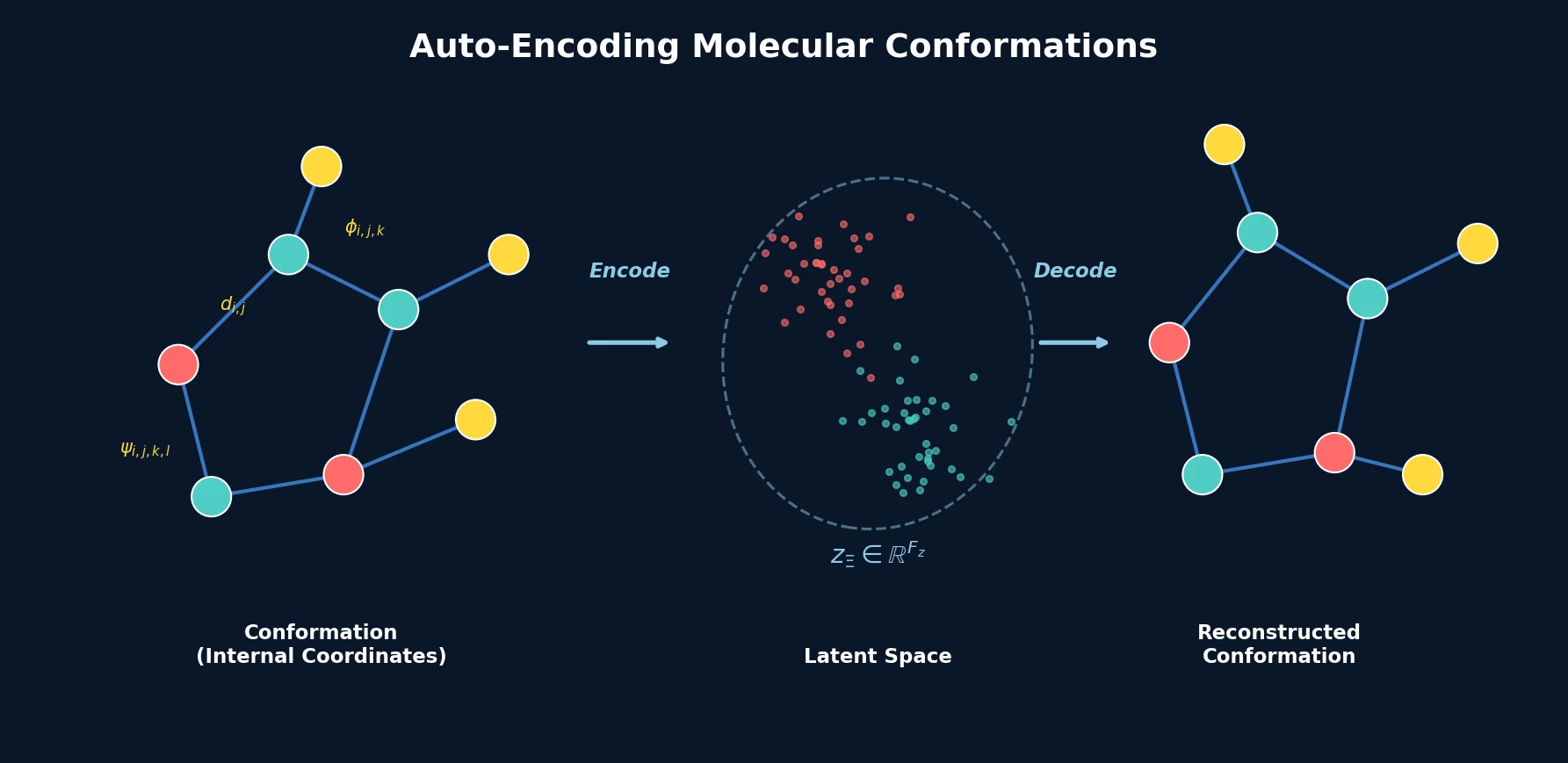
A Method for Learning Conformation Embeddings
This is a Method paper that introduces an autoencoder architecture for molecular conformations. The model converts the discrete 3D spatial arrangement of atoms (a conformation) in a given molecular graph into a continuous, fixed-size latent representation and back. The approach uses internal coordinates (bond lengths, bond angles, dihedral angles) as input rather than Cartesian coordinates, making the representation inherently invariant to rigid translations and rotations.
Why 3D Structure Matters for Molecular Modeling
Most deep learning methods for molecules operate on 2D representations: molecular graphs (atoms as nodes, bonds as edges) or SMILES strings. These representations capture connectivity and atom types but do not encode the 3D spatial arrangement of atoms. Many important molecular properties, such as the ability to fit inside a protein binding pocket or the shape-dependent pharmacological effect, depend on the molecule’s possible energetically stable spatial arrangements (conformations).
Prior work has addressed either property prediction from fixed conformations (SchNet, Schütt et al., 2018) or conformation generation for a given molecular graph (Mansimov et al., 2019; Simm and Hernández-Lobato, 2019). This paper addresses a different gap: learning a continuous, fixed-size embedding of a conformation that is independent of molecule size and atom ordering, enabling both reconstruction and generation.
Internal Coordinates and Set-Based Encoding
The core innovation is a two-part architecture: a conformation-independent graph neural network and a conformation-dependent encoder/decoder that operates on internal coordinates.
Internal Coordinate Representation
Instead of Cartesian coordinates, conformations are represented as a set of internal coordinates:
$$ \Xi = (\mathcal{D}, \Phi, \Psi) $$
where $\mathcal{D} = \{d_1, \ldots, d_{N_\mathcal{D}}\}$ are bond lengths, $\Phi = \{\phi_1, \ldots, \phi_{N_\Phi}\}$ are bond angles, and $\Psi = \{\psi_1, \ldots, \psi_{N_\Psi}\}$ are dihedral angles. This representation is invariant to rotations and rigid translations and can always be converted to and from Cartesian coordinates.
Molecular Graph Encoder
A Graph Neural Network extracts conformation-independent node embeddings from the molecular graph. The molecular graph $\mathcal{G} = (\mathcal{V}, \mathcal{E})$ uses node features $v_i \in \mathbb{R}^{F_v}$ encoding atom properties (element type, charge) and edge features $\mathbf{e}_{i,j} \in \mathbb{R}^{F_e}$ encoding bond type (single, double, triple, or aromatic). The architecture combines an edge-conditioned convolution (EConv) layer to encode bond-type information with multiple Graph Attention Network (GAT) layers:
$$ \mathbf{h}_i^l = \mathbf{GAT}^{l-1} \circ \cdots \circ \mathbf{GAT}^1 \circ \text{EConv}(\mathbf{h}_i^0) $$
where $\mathbf{h}_i^0 = v_i \in \mathbb{R}^{F_v}$ are the initial atom features. The GAT attention coefficients are:
$$ \alpha_{i,j} = \frac{\exp\left(\sigma\left(\mathbf{a}^T [\boldsymbol{\Theta}\mathbf{h}_i | \boldsymbol{\Theta}\mathbf{h}_j]\right)\right)}{\sum_{k \in \mathcal{N}(i) \cup \{i\}} \exp\left(\sigma\left(\mathbf{a}^T [\boldsymbol{\Theta}\mathbf{h}_i | \boldsymbol{\Theta}\mathbf{h}_k]\right)\right)} $$
Each GAT layer updates node embeddings using the attention weights:
$$ \mathbf{h}’_i = \alpha_{i,i}\boldsymbol{\Theta}\mathbf{h}_i + \sum_{j \in \mathcal{N}(i)} \alpha_{i,j}\boldsymbol{\Theta}\mathbf{h}_j $$
The EConv layer incorporates edge (bond-type) information via a learned filter:
$$ \mathbf{h}’_i = \boldsymbol{\Theta}\mathbf{h}_i + \sum_{j \in \mathcal{N}(i)} \mathbf{h}_j \cdot \mathrm{f}_{\boldsymbol{\Theta}}(\mathbf{e}_{i,j}) $$
where $\mathrm{f}_{\boldsymbol{\Theta}}$ is a multi-layer perceptron.
Permutation-Invariant Conformation Encoder
The conformation encoder uses a Deep Sets-style architecture (Zaheer et al., 2017) to achieve permutation invariance. Three separate neural networks encode each type of internal coordinate, conditioned on the corresponding node embeddings:
$$ z_\Xi = \frac{1}{N_\mathcal{D} + N_\Phi + N_\Psi} \left(\sum_{d \in \mathcal{D}} \rho_\Theta^{(\mathcal{D})}(\mathcal{H}, d) + \sum_{\phi \in \Phi} \rho_\Theta^{(\Phi)}(\mathcal{H}, \phi) + \sum_{\psi \in \Psi} \rho_\Theta^{(\Psi)}(\mathcal{H}, \psi)\right) $$
Each encoding function $\rho_\Theta$ takes both the internal coordinate value and the node embeddings of the involved atoms as input. The resulting conformation embedding $z_\Xi \in \mathbb{R}^{F_z}$ has a fixed dimensionality regardless of molecule size.
Conformation Decoder and Loss
Three decoder networks $\delta_\Theta^{(\mathcal{D})}$, $\delta_\Theta^{(\Phi)}$, and $\delta_\Theta^{(\Psi)}$ reconstruct internal coordinates from the conformation embedding, conditioned on the node embeddings. The reconstruction loss is:
$$ \mathcal{C}_\Xi = \frac{1}{N_\mathcal{D}} \sum_{d \in \mathcal{D}} |d - \hat{d}|_2^2 + \frac{1}{N_\Phi} \sum_{\phi \in \Phi} |\phi - \hat{\phi}|_2^2 + \frac{1}{N_\Psi} \sum_{\psi \in \Psi} \min\left(|\psi - \hat{\psi}|_2^2, 2\pi - |\psi - \hat{\psi}|_2^2\right) $$
The dihedral angle loss uses a periodic distance to account for angular periodicity. The model can be extended to a variational autoencoder (VAE) by applying the reparameterization trick from Kingma and Welling (2013).
Conformer Generation and Spatial Optimization Experiments
Dataset and Training
The model was trained on the PubChem3D dataset (Bolton et al., 2011), which contains organic molecules with up to 50 heavy atoms with multiple conformations generated by the OMEGA forcefield software.
Reconstruction Quality
Upon convergence, the model reconstructs conformations with low RMSD to the input. The median energetic difference between input and reconstructed conformations is approximately 80 kcal/mol (evaluated using the MMFF94 forcefield via RDKit), corresponding to small deviations from local minima without atom clashes.
Latent Space Structure
The learned latent space exhibits meaningful clustering: similar conformations map to nearby points, while distinct conformations separate. Principal component analysis of 200 conformations of a small molecule reveals clear conformational clusters in the first two principal components.
Conformer Generation via VAE
The variational autoencoder variant can sample diverse conformers from the learned distribution. Comparing the average inter-conformer RMSD (icRMSD) for 200 sampled conformers per molecule against the ETKDG algorithm (Riniker and Landrum, 2015) implemented in RDKit, the model achieves comparable diversity with a slightly higher average icRMSD of 0.07 Angstrom.
Multi-Objective Molecular Optimization
By combining the conformation embedding with a continuous molecular structure embedding (CDDD, Winter et al., 2019), the model enables joint optimization over both molecular graph and conformation. Using particle swarm optimization (Kennedy and Eberhart, 1995) to maximize QED (drug-likeness, values between 0 and 1) and asphericity (deviation from spherical shape, values between 0 and 1), starting from aspirin (combined score 0.76), the method finds molecules with a combined score of 1.82 after 50 iterations.
Compact Conformation Encoding with Practical Applications
The conformation autoencoder produces fixed-size latent representations of molecular 3D structures that are invariant to molecule size, atom ordering, and rigid transformations. The key findings are:
- Meaningful latent space: Conformational similarity is preserved in the embedding space, enabling clustering and interpolation.
- Diverse conformer generation: The VAE variant generates conformer ensembles with diversity comparable to established force-field-based methods.
- Joint optimization: Combining conformation and structure embeddings enables multi-objective optimization over both molecular graph and spatial arrangement.
Limitations include the relatively small energy evaluation (MMFF94 only), the lack of comparison with quantum mechanical energy evaluations, and the proof-of-concept nature of the spatial optimization experiments. The approach also relies on the quality of the internal coordinate representation, which may lose information about ring conformations and other constrained geometries.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training | PubChem3D | Multiple conformations per molecule | Organic molecules, up to 50 heavy atoms |
| Evaluation | PubChem3D holdout | Subset | Same distribution as training |
Algorithms
- Graph Neural Network: EConv + multiple GAT layers
- Conformation encoder: Deep Sets architecture with three coordinate-specific encoders
- VAE: Reparameterization trick for probabilistic sampling
- Optimization: Particle Swarm Optimization for multi-objective design
Models
- Conformation-independent: EConv + GAT layers for node embeddings
- Conformation-dependent: Three encoder/decoder feed-forward networks per coordinate type
- Latent dimension $F_z$ is fixed (exact value not specified in the workshop paper)
Evaluation
| Metric | Value | Baseline | Notes |
|---|---|---|---|
| Median energy difference | ~80 kcal/mol | Input conformations | MMFF94 forcefield |
| icRMSD difference vs ETKDG | +0.07 Angstrom | ETKDG (RDKit) | 200 conformers per molecule |
| Combined QED+asphericity | 1.82 | 0.76 (aspirin) | After 50 optimization iterations |
Hardware
Hardware details are not specified in the workshop paper.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| PubChem3D | Dataset | Public domain | NIH public database; conformations generated by OMEGA (Hawkins et al., 2010) |
| arXiv preprint | Paper | arXiv license | 6-page workshop paper, open access |
Reproducibility status: Partially Reproducible. The training dataset (PubChem3D) is publicly available, and the architecture is described in sufficient detail for reimplementation. No source code, pre-trained weights, or exact hyperparameters (latent dimension $F_z$, learning rate, number of GAT layers) are released. The workshop paper format (6 pages) limits the level of experimental detail provided.
Paper Information
Citation: Winter, R., Noé, F., & Clevert, D.-A. (2020). Auto-Encoding Molecular Conformations. Machine Learning for Molecules Workshop, NeurIPS 2020.
Publication: Machine Learning for Molecules Workshop at NeurIPS 2020
@misc{winter2021auto,
title={Auto-Encoding Molecular Conformations},
author={Winter, Robin and No\'{e}, Frank and Clevert, Djork-Arn\'{e}},
year={2021},
eprint={2101.01618},
archiveprefix={arXiv},
primaryclass={cs.LG}
}