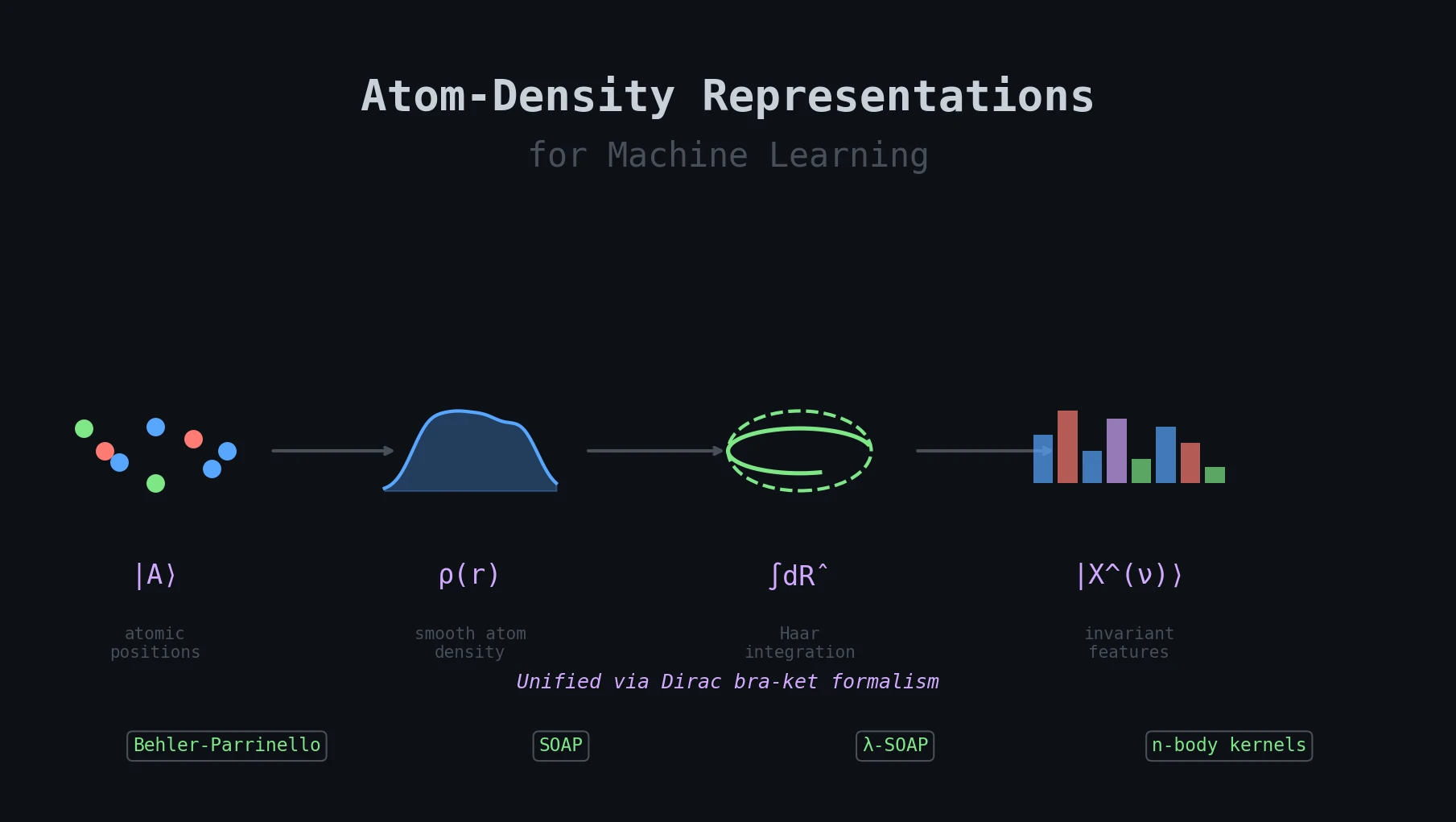
A Unified Theory of Atom-Density Representations
This is a Theory paper that provides a formal, basis-independent framework for constructing structural representations of atomic systems for machine learning. Rather than proposing a new representation, Willatt, Musil, and Ceriotti show that many popular approaches (SOAP power spectra, Behler-Parrinello symmetry functions, $n$-body kernels, and tensorial SOAP) are special cases of a single abstract construction based on smoothed atom densities and Haar integration over symmetry groups.
The Challenge of Representing Atomic Structures
Machine learning models for predicting molecular and materials properties require input representations that are (1) complete enough to distinguish structurally distinct configurations and (2) invariant to physical symmetries (translations, rotations, and permutations of identical atoms). This has led to a large and growing set of competing approaches: Coulomb matrices, symmetry functions, radial distribution functions, wavelets, invariant polynomials, and many more.
The proliferation of representations makes it difficult to compare them on equal footing or to identify which design choices are fundamental and which are incidental. Internal-coordinate approaches (e.g., Coulomb matrices) are automatically translation- and rotation-invariant but require additional symmetrization over permutations, which can introduce derivative discontinuities when done via sorting. Density-based approaches such as radial distribution functions and SOAP avoid these discontinuities by working with smooth density fields, but their theoretical connections to one another have not been made explicit.
Dirac Notation for Atomic Environments
The core innovation is to describe an atomic configuration $\mathcal{A}$ as a ket $|\mathcal{A}\rangle$ in a Hilbert space, formed by placing smooth functions $g(\mathbf{r})$ (typically Gaussians) on each atom and decorating them with orthonormal element kets $|\alpha\rangle$:
$$ \langle \mathbf{r} | \mathcal{A} \rangle = \sum_{i} g(\mathbf{r} - \mathbf{r}_{i}) | \alpha_{i} \rangle $$
This ket is basis-independent, which is the reason for adopting the Dirac notation. The same abstract object can be projected onto position space, reciprocal space, or a basis of radial functions and spherical harmonics, yielding different concrete representations that all encode the same structural information.
Symmetrization via Haar Integration
To impose translational invariance, the ket is averaged over the translation group. Averaging the raw density $|\mathcal{A}\rangle$ directly (first order, $\nu = 1$) discards all geometric information and retains only atom counts per element. The solution is to first take tensor products and then average:
$$ \left| \mathcal{A}^{(\nu)} \right\rangle_{\hat{t}} = \int \mathrm{d}\hat{t} \underbrace{\hat{t}|\mathcal{A}\rangle \otimes \hat{t}|\mathcal{A}\rangle \cdots \hat{t}|\mathcal{A}\rangle}_{\nu} $$
For $\nu = 2$, this yields a translationally-invariant ket that encodes pairwise distance information between atoms, and naturally decomposes into atom-centered contributions:
$$ \left| \mathcal{A}^{(2)} \right\rangle_{\hat{t}} = \sum_{j} |\alpha_{j}\rangle |\mathcal{X}_{j}\rangle $$
where $|\mathcal{X}_{j}\rangle$ is the environment ket centered on atom $j$, defined with a smooth cutoff function $f_{c}(r_{ij})$ that restricts each environment to a spherical neighborhood (justified by the nearsightedness principle of electronic matter). This decomposition is what justifies the widely used additive kernel between structures (a sum of kernels between environments).
Rotational Invariance and Body-Order Correlations
The same Haar integration procedure over the $SO(3)$ rotation group produces rotationally invariant representations:
$$ \left| \mathcal{X}^{(\nu)} \right\rangle_{\hat{R}} = \int \mathrm{d}\hat{R} \prod_{\aleph}^{\nu} \otimes \hat{R}\hat{U}_{\aleph}|\mathcal{X}_{j}^{\aleph}\rangle $$
The order $\nu$ of the tensor product before symmetrization determines the body-order of correlations captured: $\nu$ corresponds to $(\nu + 1)$-body correlations. The $\nu = 1$ invariant ket retains only radial (distance) information (two-body). The $\nu = 2$ ket encodes three-body correlations (two distances and an angle), and is argued to be sufficient for unique reconstruction of a configuration (up to inversion symmetry), based on extensive numerical experiments. Using nonlinear kernels (tensor products of the symmetrized ket, parameterized by $\zeta$) allows the model to incorporate higher body-order correlations beyond those explicitly in the feature vector.
Recovering SOAP, Symmetry Functions, and Tensorial Extensions
By projecting the abstract invariant kets onto specific basis sets, the authors recover several well-known frameworks as special cases.
Behler-Parrinello Symmetry Functions
In the $\delta$-function limit of the atomic density, the $\nu = 1$ and $\nu = 2$ invariant kets in real space directly correspond to the 2-body and 3-body correlation functions. Behler-Parrinello symmetry functions are projections of these correlation functions onto suitable test functions $G$:
$$ \langle \alpha \beta G_{2} | \mathcal{X}_{j} \rangle = \langle \alpha | \alpha_{j} \rangle \int \mathrm{d}r, G_{2}(r), r \left\langle \beta r | \mathcal{X}_{j}^{(1)} \right\rangle_{\hat{R},, h \to \delta} $$
where the $h \to \delta$ subscript indicates the Dirac delta limit of the atomic density.
SOAP Power Spectrum
Expanding the environmental ket in a basis of radial functions $R_{n}(r)$ and spherical harmonics $Y_{m}^{l}(\hat{\mathbf{r}})$, the $\nu = 2$ invariant ket is the SOAP power spectrum:
$$ \left\langle \alpha n \alpha’ n’ l \left| \mathcal{X}_{j}^{(2)} \right\rangle_{\hat{R}} \propto \frac{1}{\sqrt{2l+1}} \sum_{m} \langle \alpha n l m | \mathcal{X}_{j} \rangle^{\star} \langle \alpha’ n’ l m | \mathcal{X}_{j} \rangle \right. $$
This identity shows that the SOAP kernel, which can be expressed as a scalar product between truncated power spectrum vectors, is a natural consequence of the inner product between invariant kets. The $\nu = 3$ case yields the bispectrum, used as a four-body feature vector in both SOAP and Spectral Neighbor Analysis Potentials (SNAP), where its high resolution enables accurate interatomic potentials through linear regression:
$$ \langle \alpha_{1} n_{1} l_{1}, \alpha_{2} n_{2} l_{2}, \alpha n l | \mathcal{X}_{j}^{(3)} \rangle_{\hat{R}} \propto \frac{1}{\sqrt{2l+1}} \sum_{m, m_{1}, m_{2}} \langle \mathcal{X}_{j} | \alpha n l m \rangle \langle \alpha_{1} n_{1} l_{1} m_{1} | \mathcal{X}_{j} \rangle \langle \alpha_{2} n_{2} l_{2} m_{2} | \mathcal{X}_{j} \rangle \langle l_{1} m_{1} l_{2} m_{2} | l m \rangle $$
where $\langle l_{1} m_{1} l_{2} m_{2} | l m \rangle$ is a Clebsch-Gordan coefficient.
Tensorial SOAP ($\lambda$-SOAP)
The tensorial extension of SOAP incorporates an angular momentum ket $|\lambda \mu\rangle$ into the tensor product before symmetrization:
$$ \left| \mathcal{X}^{(\nu)} \lambda \mu \right\rangle_{\hat{R}} = \int \mathrm{d}\hat{R}, \hat{R}|\lambda \mu\rangle \prod_{\aleph=1}^{\nu} \otimes \hat{R}|\mathcal{X}_{j}\rangle $$
This construction is rotationally invariant in the full product space but covariant in the subspace of atomic environments, enabling models for tensorial properties (e.g., polarizability tensors, chemical shielding).
Distributions vs. Sorted Vectors
The paper also connects density-based and sorted-vector approaches. Given a set of structural descriptors ${a_{i}}$, the sorted vector is equivalent to the inverse cumulative distribution function of the histogram of values. The Euclidean distance between sorted vectors is the $\mathcal{L}^{2}$ norm of the difference between the inverse CDFs, and the $\mathcal{L}^{1}$ norm corresponds to the earth mover’s distance. This highlights that different symmetrization strategies encode essentially the same structural information.
Generalized Operators for Tuning Representations
The framework becomes especially powerful through the introduction of a linear Hermitian operator $\hat{U}$ that transforms the density ket before symmetrization. This operator must commute with rotations:
$$ \langle \alpha n l m | \hat{U} | \alpha’ n’ l’ m’ \rangle = \delta_{ll’} \delta_{mm’} \langle \alpha n l | \hat{U} | \alpha’ n’ l’ \rangle $$
Several practical modifications to standard representations can be understood as choices of $\hat{U}$:
Dimensionality Reduction
A low-rank expansion of $\hat{U}$ via PCA on the spherical-harmonic covariance matrix of environments identifies linearly independent components, enabling compression of the feature vector. For a given $l$, the covariance matrix between spherical expansion coefficients is:
$$ C_{\alpha n \alpha’ n’}^{(l)} = \frac{1}{N} \sum_{j} \sum_{m} \langle \alpha n l m | \mathcal{X}_{j} \rangle^{\star} \langle \mathcal{X}_{j} | \alpha’ n’ l m \rangle = \frac{\sqrt{2l+1}}{N} \sum_{j} \left\langle \alpha n, \alpha’ n’ l \left| \mathcal{X}_{j}^{(2)} \right\rangle_{\hat{R}} \right. $$
The eigenvectors of $\mathbf{C}^{(l)}$ provide the mixing coefficients for a compressed representation, retaining only components with significant eigenvalues.
Radial Scaling
In systems with relatively uniform atom density, the overlap kernel is dominated by the region farthest from the center. A radial scaling operator $u(r)$ (diagonal in position space) downweights distant contributions:
$$ \langle \alpha \mathbf{r} | \hat{U} | \mathcal{X}_{j} \rangle = u(r), \psi_{\mathcal{X}_{j}}^{\alpha}(\mathbf{r}) $$
This recovers the multi-scale kernels that are known to improve predictions in practice, and connects to the two-body features of Faber et al.
Alchemical Kernels
An operator that acts only in chemical-element space introduces correlations between different elements. The “alchemical” projection:
$$ \langle J \mathbf{r} | \mathcal{X}_{j} \rangle = \sum_{\alpha} u_{J\alpha}, \psi_{\mathcal{X}_{j}}^{\alpha}(\mathbf{r}) $$
reduces the dimensionality from $O(n_{\mathrm{sp}}^{2})$ to $O(d_{J}^{2})$ and has been shown to produce a low-dimensional representation of elemental space that shares similarities with periodic-table groupings.
Non-Factorizable Operators
For more complex modifications (e.g., distance- and angle-dependent scaling of three-body correlations), the operator must act on the full product space rather than factoring into independent components. The authors show that the three-body scaling function of Faber et al. corresponds to a diagonal non-factorizable operator in the real-space representation.
Implications and Future Directions
The main conclusions are:
Unification: SOAP, Behler-Parrinello symmetry functions, $\lambda$-SOAP, bispectrum descriptors, and sorted-vector approaches all emerge from the same abstract construction, differing only in the choice of basis set, body order $\nu$, and kernel power $\zeta$.
Systematic improvability: The $\hat{U}$ operator framework provides a principled way to tune representations, from simple radial scaling to full alchemical and non-factorizable couplings, with clear connections to existing heuristic modifications.
Completeness hierarchy: The body-order parameter $\nu$ and kernel power $\zeta$ together control the trade-off between completeness and computational cost. The $\nu = 2$ (three-body) representation appears to be sufficient for unique structural identification, while higher orders can be recovered through nonlinear kernels.
Limitations: The paper is primarily theoretical and does not include extensive numerical benchmarks comparing the different instantiations of the framework. Optimization of the $\hat{U}$ operator (especially in its general form) carries a risk of overfitting that the authors acknowledge but do not resolve. The connection to neural network-based representations (message-passing networks, equivariant architectures) is not explored.
Reproducibility Details
Data
This is a theoretical paper that does not introduce new benchmark results. No training or test datasets are used. The ethanol molecule in Figure 2 serves as a visualization example for three-body correlation functions.
Algorithms
The paper defines abstract constructions (Haar integration, tensor products, operator transformations) and shows how they reduce to concrete algorithms:
- SOAP power spectrum: Expand atom density in radial functions and spherical harmonics, compute $\nu = 2$ invariant ket (Eq. 33).
- Alchemical projection: Contract element channels via learned or PCA-derived mixing coefficients (Eq. 53-55).
- Dimensionality reduction: PCA on the covariance matrix $C_{\alpha n \alpha’ n’}^{(l)}$ of spherical expansion coefficients (Eq. 45).
Models
No trained models are presented. The framework applies to kernel ridge regression / Gaussian process regression models that use these representations as inputs.
Evaluation
No quantitative benchmarks are reported. The contribution is the theoretical framework itself, connecting and generalizing existing representations.
Hardware
Not applicable (theoretical work).
Paper Information
Citation: Willatt, M. J., Musil, F., & Ceriotti, M. (2019). Atom-density representations for machine learning. The Journal of Chemical Physics, 150(15), 154110. https://doi.org/10.1063/1.5090481
Publication: The Journal of Chemical Physics, 2019
@article{willatt2019atom,
title={Atom-density representations for machine learning},
author={Willatt, Michael J. and Musil, F{\'e}lix and Ceriotti, Michele},
journal={The Journal of Chemical Physics},
volume={150},
number={15},
pages={154110},
year={2019},
doi={10.1063/1.5090481},
eprint={1807.00408},
archiveprefix={arXiv},
primaryclass={physics.chem-ph}
}