Overview
SELFIES (SELF-referencIng Embedded Strings) is a string-based molecular representation where every possible string, even one generated randomly, corresponds to a syntactically and semantically valid molecule. This property addresses a major limitation of SMILES, where a large fraction of strings produced by machine learning models represent invalid chemical structures.
The format is implemented in an open-source Python library called selfies. Since the original publication, the library has undergone significant architectural changes, most notably replacing the original string-manipulation engine with a graph-based internal representation that improved both performance and extensibility (see Recent Developments).
Key Characteristics
- Guaranteed Validity: Every possible SELFIES string can be decoded into a valid molecular graph that obeys chemical valence rules. This is its fundamental advantage over SMILES.
- Machine Learning Friendly: Can be used directly in any machine learning model (like VAEs or GANs) without adaptation, guaranteeing that all generated outputs are valid molecules.
- Customizable Constraints: The underlying chemical rules, such as maximum valence for different atoms, can be customized by the user. The library provides presets (e.g., for hypervalent species) and allows users to define their own rule sets.
- Human-readable: With some familiarity, SELFIES strings are human-readable, allowing interpretation of functional groups and connectivity.
- Local Operations: SELFIES encodes branch length and ring size as adjacent symbols in the string (rather than requiring matched delimiters or repeated digits at distant positions, as SMILES does), preventing common syntactical errors like unmatched parentheses or mismatched ring-closure digits.
- Broad Support: The current
selfieslibrary supports aromatic molecules (via kekulization), isotopes, charges, radicals, and stereochemistry. It also includes a dot symbol (.) for representing disconnected molecular fragments.
Basic Syntax
SELFIES uses symbols enclosed in square brackets (e.g., [C], [O], [#N]). The interpretation of each symbol depends on the current state of the derivation (described below), which ensures chemical valence rules are strictly obeyed. The syntax is formally defined by a Chomsky type-2 context-free grammar.
Derivation Rules
SELFIES are constructed using a table of derivation rules. The process starts in an initial state (e.g., $X_0$) and reads the SELFIES string symbol by symbol. Each symbol, combined with the current state, determines the resulting atom/bond and the next state. The derivation state $X_n$ intuitively tracks that the previously added atom can form a maximum of $n$ additional bonds.
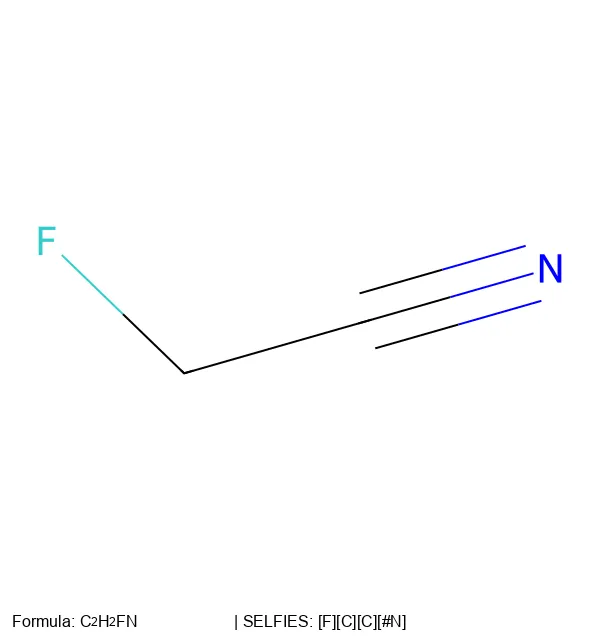
For example, the string [F][=C][=C][#N] is derived as follows, where $X_n$ indicates the atom can form up to $n$ additional bonds. Notice how bond demotion occurs: the first [=C] requests a double bond, but only a single bond is formed because state $X_1$ limits the connection to one bond.
$$ \begin{aligned} \text{State } X_0 + \text{[F]} &\rightarrow \text{F} + \text{State } X_1 \\ \text{State } X_1 + \text{[=C]} &\rightarrow \text{F-C} + \text{State } X_3 \\ \text{State } X_3 + \text{[=C]} &\rightarrow \text{F-C=C} + \text{State } X_2 \\ \text{State } X_2 + [\#\text{N}] &\rightarrow \text{F-C=C=N} + \text{Final} \end{aligned} $$
Structural Features
- Branches: Represented by a
[Branch]symbol. The symbols immediately following it are interpreted as an index that specifies the number of SELFIES symbols belonging to that branch. This structure prevents errors like unmatched parentheses in SMILES. - Rings: Represented by a
[Ring]symbol. Similar to branches, subsequent symbols specify an index that indicates which previous atom to connect to, forming a ring closure. To avoid violating valence constraints, ring bond creation is postponed to a final post-processing step, where it is only completed if the target atom has available bonds.
Examples
To see how these derivation rules work in practice, here are SELFIES representations for common molecules of increasing complexity:

[C][C][O]
[C][=C][C][=C][C][=C][Ring1][=Branch1]
[C][C][Branch1][C][=O][O][C][=C][C][=C][C][=C][Ring1][=Branch1][C][Branch1][C][=O][O]The selfies Python Library
The selfies library provides a dependency-free Python implementation. Here are the core operations:
import selfies as sf
# SMILES -> SELFIES
smiles = "c1ccc(C(=O)O)cc1" # benzoic acid
encoded = sf.encoder(smiles)
print(encoded)
# -> [C][=C][C][=C][C][Branch1][C][=O][O][=C][Ring1][=Branch1]
# SELFIES -> SMILES
decoded = sf.decoder(encoded)
print(decoded)
# -> C1=CC=CC(=C1)C(=O)O
# Robustness: random strings always decode to valid molecules
random_selfies = "[C][F][Ring1][O][=N][Branch1][C][S]"
print(sf.decoder(random_selfies))
# -> always returns a valid molecule
Tokenization and Encoding
import selfies as sf
selfies_str = "[C][=C][C][=C][C][Branch1][C][=O][O][=C][Ring1][=Branch1]"
# Tokenize into individual symbols
tokens = list(sf.split_selfies(selfies_str))
print(tokens)
# -> ['[C]', '[=C]', '[C]', '[=C]', '[C]', '[Branch1]', '[C]',
# '[=O]', '[O]', '[=C]', '[Ring1]', '[=Branch1]']
# Get the alphabet (unique token set) from a dataset
dataset = ["[C][C][O]", "[C][=C][C][=C][C][=C][Ring1][=Branch1]"]
alphabet = sf.get_alphabet_from_selfies(dataset)
print(sorted(alphabet))
# -> ['[=Branch1]', '[=C]', '[C]', '[O]', '[Ring1]']
# Convert to integer encoding for ML pipelines
encoding, _ = sf.selfies_to_encoding(
selfies=selfies_str,
vocab_stoi={s: i for i, s in enumerate(sorted(alphabet))},
pad_to_len=20,
enc_type="label",
)
Customizing Valence Constraints
import selfies as sf
# View current constraints
print(sf.get_semantic_constraints())
# Allow hypervalent sulfur (e.g., SF6)
sf.set_semantic_constraints("hypervalent")
# Or define custom constraints
sf.set_semantic_constraints({
"S": 6, # allow hexavalent sulfur
"P": 5, # allow pentavalent phosphorus
})
# Reset to defaults
sf.set_semantic_constraints("default")
SELFIES in Machine Learning
Molecular Generation
SELFIES is particularly advantageous for generative models in computational chemistry. When used in a VAE, the entire continuous latent space decodes to valid molecules, unlike SMILES where large regions of the latent space are invalid. The original SELFIES paper demonstrated this concretely: a VAE trained with SELFIES stored two orders of magnitude more diverse molecules than a SMILES-based VAE, and a GAN produced 78.9% diverse valid molecules compared to 18.6% for SMILES (Krenn et al., 2020).
Several generation approaches build directly on SELFIES:
- Latent space optimization: LIMO uses a SELFIES-based VAE with gradient-based optimization to generate molecules with nanomolar binding affinities, achieving 6-8x speedup over RL baselines (Eckmann et al., 2022).
- Training-free generation: STONED demonstrates that simple character-level mutations in SELFIES (replacement, deletion, insertion) produce valid molecules by construction, eliminating the need for neural networks entirely. STONED achieved a GuacaMol score of 14.70, competitive with deep generative models (Nigam et al., 2021).
- Gradient-based dreaming: PASITHEA computes gradients with respect to one-hot encoded SELFIES inputs to steer molecules toward target property values. Because SELFIES’ surjective mapping guarantees every intermediate representation is a valid molecule, this continuous optimization over the input space is feasible. PASITHEA generated molecules with properties outside the training data range (logP up to 4.24 vs. a training max of 3.08), with 97.2% novelty (Shen et al., 2021).
- Large-scale pre-training: MolGen is a BART-based model pre-trained on 100M+ SELFIES molecules. It achieves 100% validity and an FCD of 0.0015 on MOSES (vs. 0.0061 for Chemformer), and introduces chemical feedback to align outputs with preference rankings (Fang et al., 2024).
In benchmarks, SELFIES performs well for optimization-oriented tasks. In the PMO benchmark of 25 methods, SELFIES-REINVENT ranked 3rd and STONED ranked 5th. SELFIES-based genetic algorithms outperformed SMILES-based GAs, likely because SELFIES provides more intuitive mutation operations (Gao et al., 2022). The Tartarus benchmark corroborates this across more diverse real-world objectives (organic emitters, protein ligands, reaction substrates): SELFIES-VAE consistently outperforms SMILES-VAE, and the representation matters most where validity is a bottleneck (Nigam et al., 2022).
SELFIES mutations provide a simple but effective way to explore chemical space:
import selfies as sf
import random
def mutate_selfies(selfies_str, mutation_type="replace"):
"""Mutate a SELFIES string. Every output is a valid molecule."""
tokens = list(sf.split_selfies(selfies_str))
alphabet = list(sf.get_semantic_robust_alphabet())
idx = random.randint(0, len(tokens) - 1)
if mutation_type == "replace":
tokens[idx] = random.choice(alphabet)
elif mutation_type == "insert":
tokens.insert(idx, random.choice(alphabet))
elif mutation_type == "delete" and len(tokens) > 1:
tokens.pop(idx)
return "".join(tokens)
# Every mutation produces a valid molecule
original = sf.encoder("c1ccccc1") # benzene
for _ in range(5):
mutant = mutate_selfies(original)
print(sf.decoder(mutant)) # always valid
Property Prediction and Pretraining
SELFormer is a RoBERTa-based chemical language model pretrained on 2M ChEMBL compounds using SELFIES as input. Because every masked token prediction corresponds to a valid molecular fragment, the model never wastes capacity learning invalid chemistry. SELFormer outperformed ChemBERTa-2 by approximately 12% on average across BACE, BBBP, and HIV classification benchmarks (Yüksel et al., 2023). ChemBERTa also evaluated SELFIES as an input representation, finding comparable performance to SMILES on the Tox21 task (Chithrananda et al., 2020).
The Regression Transformer demonstrated that SELFIES achieves ~100% validity vs. ~40% for SMILES in conditional molecular generation, while performing comparably for property prediction. This dual prediction-generation capability is enabled by interleaving numerical property tokens with SELFIES molecular tokens in a single sequence (Born & Manica, 2023).
At larger scales, ChemGPT (up to 1B parameters) uses a GPT-Neo backbone with SELFIES tokenization for autoregressive molecular generation, demonstrating that SELFIES follows the same power-law neural scaling behavior observed in NLP (Frey et al., 2023).
Optical Chemical Structure Recognition
In image-to-text chemical structure recognition, Rajan et al. (2022) compared SMILES, DeepSMILES, SELFIES, and InChI as output formats using the same transformer architecture. SELFIES achieved 100% structural validity (every prediction could be decoded), while SMILES predictions occasionally contained syntax errors. The trade-off: SMILES achieved higher exact match accuracy (88.62%) partly because SELFIES strings are longer, producing more tokens for the decoder to predict.
Chemical Name Translation
STOUT uses SELFIES as its internal representation for translating between chemical line notations and IUPAC names. All SMILES are converted to SELFIES before processing, and the model achieves a BLEU score of 0.94 for IUPAC-to-SELFIES translation and 0.98 Tanimoto similarity on valid outputs. The authors found SELFIES’ syntactic robustness particularly valuable for this sequence-to-sequence task, where the decoder must produce a chemically valid output string (Rajan et al., 2021).
Tokenization
Converting SELFIES strings into tokens for neural models is more straightforward than SMILES tokenization. Each bracket-enclosed symbol ([C], [=C], [Branch1]) is a natural token boundary. Atom Pair Encoding (APE) extends byte pair encoding with chemistry-aware constraints for both SMILES and SELFIES. For SELFIES specifically, APE preserves atomic identity during subword merging, and SELFIES models showed strong inter-tokenizer agreement: all true positives from SELFIES-BPE were captured by SELFIES-APE (Leon et al., 2024).
Limitations and Trade-offs
Validity Constraints Can Introduce Bias
The guarantee that every string decodes to a valid molecule is SELFIES’ core advantage, but recent work has shown this comes with trade-offs. Skinnider (2024) demonstrated that SMILES-based models consistently outperform SELFIES-based models on distribution-learning tasks. The mechanism: invalid SMILES represent a model’s least confident predictions, and filtering them out acts as implicit quality control. SELFIES models, by construction, cannot discard low-confidence outputs this way. Furthermore, SELFIES validity constraints introduce systematic structural biases, generating fewer aromatic rings and more aliphatic structures compared to training data. When SELFIES constraints were relaxed to allow invalid generation (“unconstrained SELFIES”), performance improved, providing causal evidence that the ability to generate and discard invalid outputs benefits distribution learning.
This finding reframes the SMILES vs. SELFIES choice as context-dependent. As Grisoni (2023) summarizes in a review of chemical language models: “SMILES offer a richer, more interpretable language with well-studied augmentation strategies, while SELFIES guarantee validity at the cost of chemical realism and edit interpretability.”
The PMO benchmark provides further nuance: SELFIES-based variants of language model methods (REINVENT, LSTM HC, VAE) generally do not outperform their SMILES counterparts, because modern language models learn SMILES grammar well enough that syntactic invalidity is no longer a practical bottleneck. The exception is genetic algorithms, where SELFIES mutations are naturally well-suited.
A study on complex molecular distributions paints a consistent picture: SELFIES-trained RNNs achieve better standard metrics (validity, uniqueness, novelty), while SMILES-trained RNNs achieve better distributional fidelity as measured by Wasserstein distance (Flam-Shepherd et al., 2022). Taken together, these findings suggest that SELFIES and SMILES have genuinely complementary strengths, and the best choice depends on whether the task prioritizes validity/novelty or distributional faithfulness.
Degenerate Outputs
Although every SELFIES string decodes to a valid molecule, the decoded molecule may not always be chemically meaningful in context. The Regression Transformer reported ~1.9% defective generations where the output molecule had fewer than 50% of the seed molecule’s atoms (Born & Manica, 2023). This highlights a distinction between syntactic validity (which SELFIES guarantees) and semantic appropriateness (which it does not).
Other Limitations
- Indirect Canonicalization: A canonical SELFIES string is currently generated by first creating a canonical SMILES string and then converting it to SELFIES. Direct canonicalization is a goal for future development.
- String Length: SELFIES strings are generally longer than their corresponding SMILES strings, which can impact storage, processing times, and sequence modeling difficulty for very large datasets.
- Ongoing Standardization: While the library now supports most major features found in SMILES, work is ongoing to extend the format to more complex systems like polymers, crystals, and reactions.
Variants and Extensions
Group SELFIES
Group SELFIES extends the representation with group tokens that represent functional groups or entire substructures (e.g., a benzene ring or carboxyl group) as single units. Each group token has labeled attachment points with specified valency, allowing the decoder to continue tracking available bonds. Group SELFIES maintains the validity guarantee while producing shorter, more human-readable strings. On MOSES VAE benchmarks, Group SELFIES achieved an FCD of 0.1787 versus 0.6351 for standard SELFIES, indicating substantially better distribution learning (Cheng et al., 2023).
STONED Algorithms
STONED (Superfast Traversal, Optimization, Novelty, Exploration and Discovery) is a suite of algorithms that exploit SELFIES’ validity guarantee for training-free molecular design through point mutations, interpolation, and optimization (Nigam et al., 2021). See Molecular Generation above for benchmark results.
Recent Developments
The 2023 library update replaced the original string-manipulation engine with a graph-based internal representation. This change resolved several long-standing limitations: the original approach could not handle aromatics (requiring kekulization), stereochemistry, or charged species. The graph-based engine now supports all of these, and processes 300K+ molecules in approximately 4 minutes in pure Python. The library has been validated on all 72 million molecules from PubChem.
Looking forward, researchers have outlined 16 future research directions for extending robust representations to complex systems like polymers, crystals, and chemical reactions.
Further Reading
- Converting SELFIES Strings to 2D Molecular Images: Hands-on tutorial demonstrating SELFIES robustness and building visualization tools
References
- Krenn, M., Häse, F., Nigam, A., Friederich, P., & Aspuru-Guzik, A. (2020). Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Machine Learning: Science and Technology, 1(4), 045024.
- Krenn, M., Ai, Q., Barthel, S., Carson, N., Frei, A., Frey, N. C., … & Aspuru-Guzik, A. (2022). SELFIES and the future of molecular string representations. Patterns, 3(10), 100588.
- Lo, A., Pollice, R., Nigam, A., White, A. D., Krenn, M., & Aspuru-Guzik, A. (2023). Recent advances in the self-referencing embedded strings (SELFIES) library. Digital Discovery, 2, 897-908.
- Skinnider, M. A. (2024). Invalid SMILES are beneficial rather than detrimental to chemical language models. Nature Machine Intelligence, 6, 437-448.
- Shen, C., Krenn, M., Eppel, S., & Aspuru-Guzik, A. (2021). Deep molecular dreaming: inverse machine learning for de-novo molecular design and interpretability with surjective representations. Machine Learning: Science and Technology, 2(3), 03LT02.
- Fang, Y., et al. (2024). Domain-agnostic molecular generation with chemical feedback. ICLR 2024.
- Born, J., & Manica, M. (2023). Regression Transformer enables concurrent sequence regression and generation for molecular language modelling. Nature Machine Intelligence, 5, 432-444.
- Frey, N. C., Soklaski, R., Axelrod, S., Samsi, S., Gómez-Bombarelli, R., Coley, C. W., & Gadepally, V. (2023). Neural scaling of deep chemical models. Nature Machine Intelligence, 5, 1297-1305.
- Rajan, K., Zielesny, A., & Steinbeck, C. (2021). STOUT: SMILES to IUPAC names using neural machine translation. Journal of Cheminformatics, 13, 34.
- Nigam, A., Pollice, R., & Aspuru-Guzik, A. (2022). Tartarus: A benchmarking platform for realistic and practical inverse molecular design. NeurIPS 2022 Datasets and Benchmarks.
- SELFIES GitHub Repository