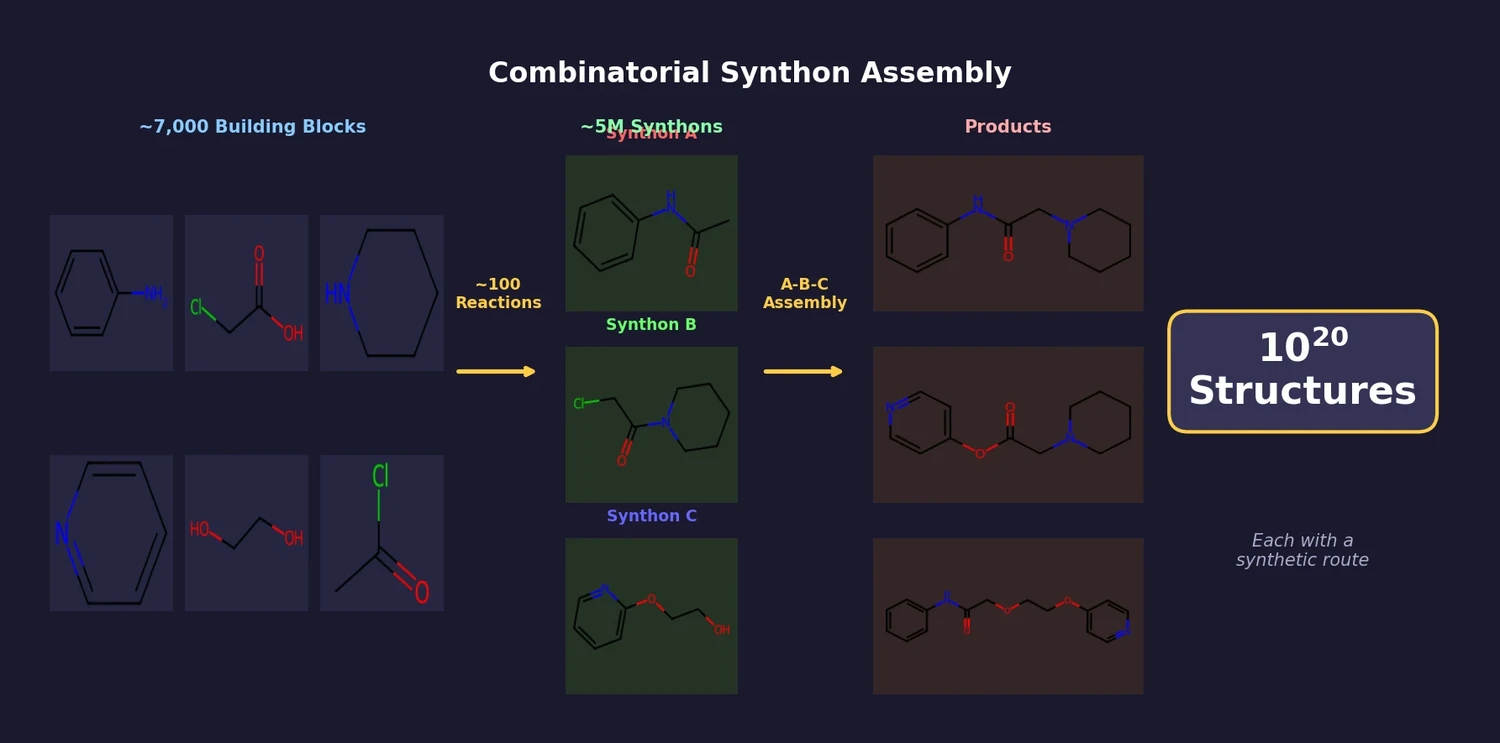
Combinatorial Synthon Assembly at Scale
AllChem is a computer-aided molecular design system that generates and searches an unprecedentedly large space of synthetically accessible structures (on the order of $10^{20}$). Rather than enumerating molecules from mathematical graphs (as in the GDB databases), AllChem builds its chemical space from real synthetic chemistry: it recursively applies known reactions to commercial building blocks, producing synthons (structures with open valences of defined reactivity) that combinatorially assemble into complete molecules. Every structure found by a search comes paired with a proposed synthetic route.
Motivation: Costs and Benefits Together
Most computer-aided molecular design methods focus on predicting biological activity (the benefit) while leaving synthesis feasibility (the cost) to the laboratory chemist. AllChem addresses both simultaneously. Its predecessor, ChemSpace, accessed $\sim 10^{14}$ structures built from simple combinatorial libraries (chemist-proposed scaffolds plus commercial side chains), but only about 5% of structures in the medicinal chemistry literature fit that template. AllChem aims to cover roughly 50% of published structures by allowing multi-step synthon generation that produces more complex, non-trivial scaffolds.
The gensyn Synthon Generator
The core component is gensyn, a program that recursively applies a curated set of approximately 100 reactions to approximately 7,000 commercially available building blocks. Each product becomes a new building block for subsequent reaction steps, with recursion bounded primarily by a cumulative synthesis “cost” limit (roughly five AllChem-type steps per sequence). Structures bearing open valences are collected as synthons. A typical run produces around $5 \times 10^6$ synthons, which combinatorially represent $(5 \times 10^6)^3 = 10^{20}$ complete structures with an A-B-C topology.
Key design decisions in gensyn:
- Reaction curation: All reactions come from external human-readable text files, based on reactions already practiced by laboratory chemists. Scope constraints are calibrated so that at least 90% of randomly sampled reaction applications appear unchallengeable to synthetic chemists.
- Reactive intermediates: Explicitly represented. For example, amide formation requires three steps: acid chloride to electrophilic synthon, amine to nucleophilic synthon, then coupling.
- Protective groups: Addition and removal are treated as standard reactions.
- Concerted cyclizations: Represented by splitting the ring formation across two complementary synthons with specially labeled open valences.
- Bimolecular reactions: In addition to unimolecular transformations, gensyn performs reactions that combine selected synthons with other synthons, increasing overall structural diversity.
- Constraints: Maximum of one prochiral center (to avoid diastereomeric mixtures), heavy atom count limits for lead-likeness, and a cumulative cost bound on synthetic routes. Each reaction step has a default cost of $-5$, and the maximum allowed cumulative cost is $-25$ (roughly five steps per sequence).
Reaction Description Language
Reactions are described using an extension of Sybyl Line Notation (SLN), a SMILES-like notation. Each reaction description specifies the structural pattern required in the substrate, the transformation to apply, the reactivity class of resulting open valences, the relative cost, incompatible functional groups, and rules for handling multiple equivalent reactive sites. A separate reactivity table defines which valence classes can react with each other (e.g., nucleophilic with electrophilic).
Topomer Similarity Search
Searching among $10^{20}$ complete structures relies on topomer shape similarity as a branch-and-bound filter. A query structure is fragmented by breaking acyclic single bonds (individually and pairwise), each fragment is converted to a topomer (a canonical 3D shape), and the topomer is compared against all stored synthons. Topomer comparisons run at tens of thousands per second. Because the vast majority of synthons are individually shape-dissimilar enough to eliminate every complete structure containing them, the search space collapses rapidly. To be acceptable, a product must also have been formed by joining open valences with complementary reactivity.
Validation used repeated “self-searches,” in which a query structure is assembled from randomly chosen synthons and searched for in the database. On the 250,000-synthon leadhopping database, average self-search time was 7.1 minutes; complete searches of the full-scale database take several hours on standard hardware.
Applications: Lead Hopping and Scaffold Generation
Lead hopping: Finding structurally novel molecules that are shape-similar (and therefore likely biologically similar) to a query lead. Using a 250,000-synthon leadhopping database, 18 of 19 self-search queries recovered the query structure perfectly (shape difference of 0 topomer units). The remaining query also recovered itself as the closest hit.
Scaffold idea generation: Filtering the synthon collection for small ($\leq$ 14 heavy atoms), low-chirality scaffolds with at least two diversification sites (primarily through nucleophilic heteroatom reactions on activated carbon electrophiles or Suzuki-type couplings), UV chromophores, minimal freely rotatable bonds (especially between diversification sites and rings), a ring, and short synthetic paths (all branches fewer than about six AllChem steps). Over 20% of gensyn-proposed synthons pass these scaffold filters, suggesting on the order of $10^6$ accessible and structurally distinct scaffolds, compared to the few thousand scaffolds typically represented in large screening collections.
Compute and Infrastructure
Full-scale synthon database recreation takes approximately one week using two standard workstations (one Oracle database server, one compute engine). The codebase was rewritten from Java to Python for portability and performance. All data is managed through an Oracle relational database, including synthons, intermediates, and a reactions table recording every gensyn conversion.
Limitations
- Variable reactivity of open valences (e.g., weakly nucleophilic amines may not form the implied bond readily) is handled only approximately via reagent class annotations.
- Stereospecificity and most aromatic electrophilic substitution reactions are omitted.
- The system was described as under active development at the time of publication, giving the paper the character of an interim progress report.
- Drug-likeness of 3-synthon products (average MW ~800, CLOGP ~8.0) requires careful filtering of the synthon distribution toward smaller, less lipophilic components.
Reproducibility Details
AllChem was developed as proprietary software at Tripos Inc. (Tripos Discovery Research, Bude, Cornwall, UK). No source code, synthon databases, or reaction files have been publicly released. The paper functions as a description of the system’s architecture and early results rather than a reproducibility-oriented publication.
- Code: Not publicly available. The system was proprietary to Tripos Inc.
- Data: Synthon databases and reaction description files are not shared.
- Hardware: Two standard workstations (one Oracle server, one compute engine); no specialized hardware required.
- Funding: NIH/GMS SBIR grant 2 R44 GM068359-02.
Reproducibility status: Closed.
Paper Information
- Journal: Journal of Computer-Aided Molecular Design, Vol. 21, No. 6, pp. 341-350
- Published: January 25, 2007
@article{cramer2007allchem,
title={AllChem: generating and searching 10^{20} synthetically accessible structures},
author={Cramer, Richard D. and Soltanshahi, Farhad and Jilek, Robert J. and Campbell, Brian},
journal={Journal of Computer-Aided Molecular Design},
volume={21},
number={6},
pages={341--350},
year={2007},
publisher={Springer Science+Business Media},
doi={10.1007/s10822-006-9093-8}
}