Diversity-Biased Search of the Small Molecule Universe
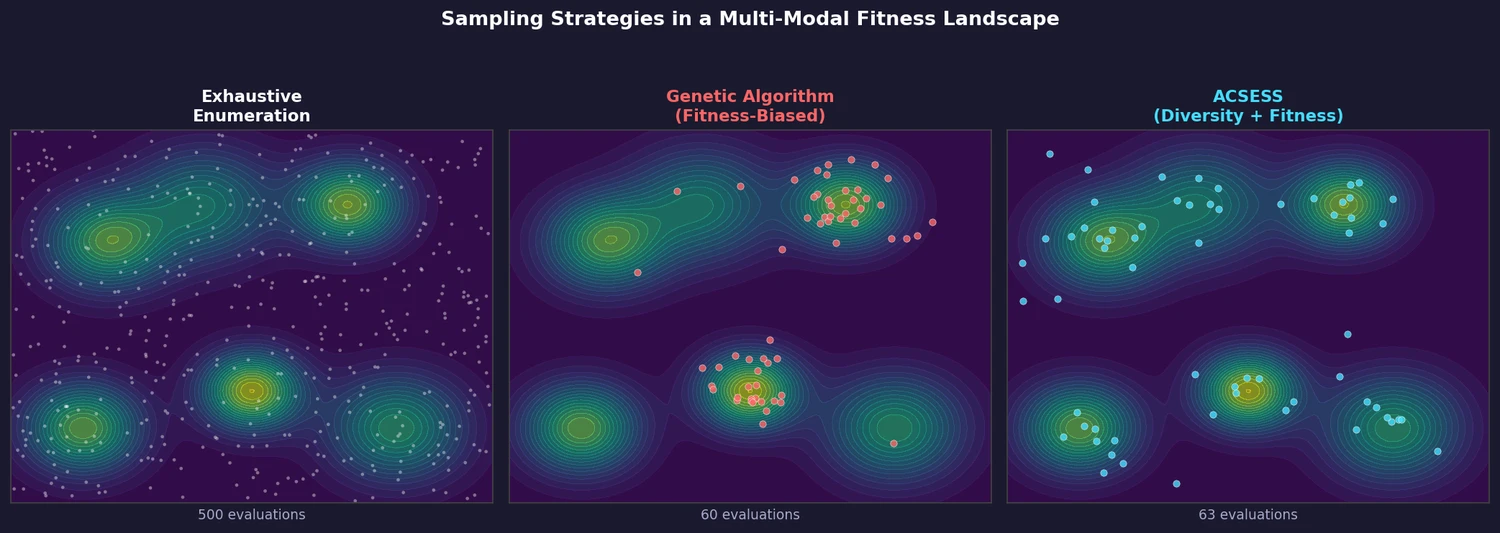
The small molecule universe (SMU), estimated at over $10^{60}$ synthetically feasible organic molecules under ~500 Da, is far too large for exhaustive enumeration and evaluation. This paper extends the ACSESS (Algorithm for Chemical Space Exploration with Stochastic Search) framework to simultaneously optimize molecular diversity and a targeted physical property. The key insight is that enforcing diversity at each iteration prevents the search from collapsing into local optima, a failure mode common in standard genetic algorithms.
Motivation: Diversity vs. Fitness
Standard genetic algorithms optimize fitness effectively but sacrifice diversity: they converge to a few high-fitness regions while ignoring equally good solutions elsewhere. Exhaustive enumeration guarantees completeness but is computationally infeasible beyond ~20 heavy atoms. ACSESS bridges this gap by maintaining a maximally diverse library throughout the optimization process, ensuring coverage of multiple fitness peaks without needing to evaluate every candidate.
The Property-Optimizing ACSESS Algorithm
The method has four iterative steps:
- Initialize a library (from a single molecule or a seed collection)
- Breed new compounds via mutations and crossovers
- Filter by property threshold, removing compounds below a cutoff
- Select a maximally diverse subset of qualifying structures
The property threshold increases linearly with each iteration, starting low (to prevent population collapse) and gradually rising until the desired fitness level is reached. Diversity is enforced via either a maximin algorithm (maximizing nearest-neighbor distance) or cell-based partitioning (linear scaling for large libraries).
Molecules are represented in a 40-dimensional chemical space using Moreau-Broto autocorrelation descriptors. The descriptor encodes correlations of atomic properties as a function of topological distance (bond distance) $d$:
$$ AC(d, p) = \sum_{i \leq j} p_{i} , p_{j} , \delta(d_{ij} - d) $$
where $p_{i}$ is an atomic property of atom $i$ and $d_{ij}$ is the shortest bond path between atoms $i$ and $j$. Five atomic properties are used: atomic number, Gasteiger-Marsili partial charge, atomic polarizability, topological steric index, and unity ($p_{i} = 1$ for all $i$, effectively counting atom pairs at each distance). Topological distance $d$ ranges from 0 to 7, yielding $5 \times 8 = 40$ descriptor components. Descriptors are mean-centered and normalized to unit variance before computing distances.
Chemical space distance is the Euclidean distance between descriptor vectors:
$$ D_{ij} = \sqrt{\sum_{k=1}^{N} (d_{ik} - d_{jk})^2} $$
Library diversity is measured as the average nearest-neighbor distance:
$$ D_{\min} = \frac{1}{M} \sqrt{\sum_{i=1}^{M} \min_{i \neq j} (D_{ij}^2)} $$
Validation on NKp Fitness Landscapes
The NKp model maps binary strings of length $N$ to fitness values in $[0, 1]$. The fitness of a string $g$ is:
$$ \Phi(g) = \frac{1}{N} \sum_{i=1}^{N} \varphi_{i}(g) $$
where each $\varphi_{i} \in [0, 1]$ is a randomly drawn fitness contribution. Ruggedness is controlled by $K$ (the number of inter-bit associations per position) and $p$ (fitness contribution weights). Using $N = 19$, $K = 9$, $p = 0.9$ (524,288 total strings, comparable to GDB-9 size), the global maximum was ~0.3. Both ACSESS and SGA were initialized with the same diverse subset and ran for 30 iterations across 10 independent runs:
- ACSESS found the global optimum in 100% of runs (vs. 60% for SGA)
- ACSESS discovered ~15 of 19 globally optimal strings on average (vs. ~3 for SGA)
- ACSESS solutions had higher average fitness than SGA solutions
Validation on GDB-9 Dipole Moments
The method was tested on all ~300,000 molecules in GDB-9 (up to 9 heavy atoms; allowed atom types: C, N, O, S, Cl). For each molecule, the Boltzmann-averaged dipole moment was computed at the AM1 level (Gaussian 09):
$$ D = \frac{\sum_{i \in C} \mu_{i} , e^{-\beta E_{i}}}{\sum_{i \in C} e^{-\beta E_{i}}} $$
where $\mu_{i}$ and $E_{i}$ are the dipole moment and internal energy of conformation $i$, and $\beta = 1 / (k_{\text{B}} T)$ at $T = 298$ K. Conformations (including stereoisomers) were generated using OpenEye OMEGA. The target was molecules with dipole moments $\geq 5.5$ D (the 90th percentile). ACSESS first generated a maximally diverse seed set, then ran 60 iterations of fitness-biased optimization. All methods were initialized from the same diverse seed and compared over multiple runs.
| Method | Dipole Moment (D) | Diversity (eq. 4) |
|---|---|---|
| GA-Roulette | 5.8 $\pm$ 0.03 | 6.5 $\pm$ 0.7 |
| GA-Tournament | 6.4 $\pm$ 0.08 | 3.5 $\pm$ 0.7 |
| GA-Elitism | 6.74 $\pm$ 0.08 | 5.4 $\pm$ 0.4 |
| ACSESS | 6.05 $\pm$ 0.05 | 9.7 $\pm$ 0.6 |
ACSESS achieved nearly double the diversity of the best SGA variant while maintaining competitive fitness. Its diversity (~9.7) approached the diversity of the full enumerated high-fitness subset of GDB-9 (~12). Self-organizing map (SOM) visualizations confirmed that ACSESS covered high-activity regions that SGAs missed entirely.
Only ~30,000 fitness evaluations were needed to locate diverse optimal regions in the 300,000-molecule space, a 10x efficiency gain over exhaustive enumeration.
Limitations
- Tested only on relatively small chemical spaces (GDB-9 with ~300k molecules and 19-bit NKp with ~500k strings); scaling to the full SMU ($10^{60}$) remains a research direction
- Property evaluation (AM1 dipole moments with conformer generation) is the computational bottleneck, not the ACSESS algorithm itself
- The 40-dimensional autocorrelation descriptor space may not capture all relevant structural features for every optimization target
- Comparison is limited to simple genetic algorithms; more sophisticated evolutionary strategies were not benchmarked
Reproducibility Details
The ACSESS algorithm relies on proprietary software, limiting full reproducibility.
| Artifact | Type | License | Notes |
|---|---|---|---|
| GDB-9 | Dataset | CC-BY-4.0 | Publicly available enumerated chemical universe (~300k molecules) |
- Code: No public source code was released. The implementation depends on OpenEye OEChem TK (molecule generation), OpenEye MolProp TK (filtering), and OpenEye OMEGA TK (conformer generation), all of which require commercial licenses.
- Property calculations: Dipole moments were computed at the AM1 level using Gaussian 09, also commercial software.
- NKp landscape: Fully specified by parameters ($N = 19$, $K = 9$, $p = 0.9$) and standard NKp model equations, making this portion independently reproducible.
- Hardware: No specific compute requirements reported.
- Reproducibility status: Partially Reproducible. The algorithm is well-described and the NKp experiments could be reimplemented, but the molecular experiments require OpenEye and Gaussian 09 licenses, and no reference implementation was released.
Paper Information
- Journal: Journal of Chemical Information and Modeling, Vol. 55, No. 3, pp. 529-537
- Published: January 16, 2015
@article{rupakheti2015strategy,
title={Strategy To Discover Diverse Optimal Molecules in the Small Molecule Universe},
author={Rupakheti, Chetan and Virshup, Aaron M. and Yang, Weitao and Beratan, David N.},
journal={Journal of Chemical Information and Modeling},
volume={55},
number={3},
pages={529--537},
year={2015},
publisher={American Chemical Society},
doi={10.1021/ci500749q}
}