Key Contribution
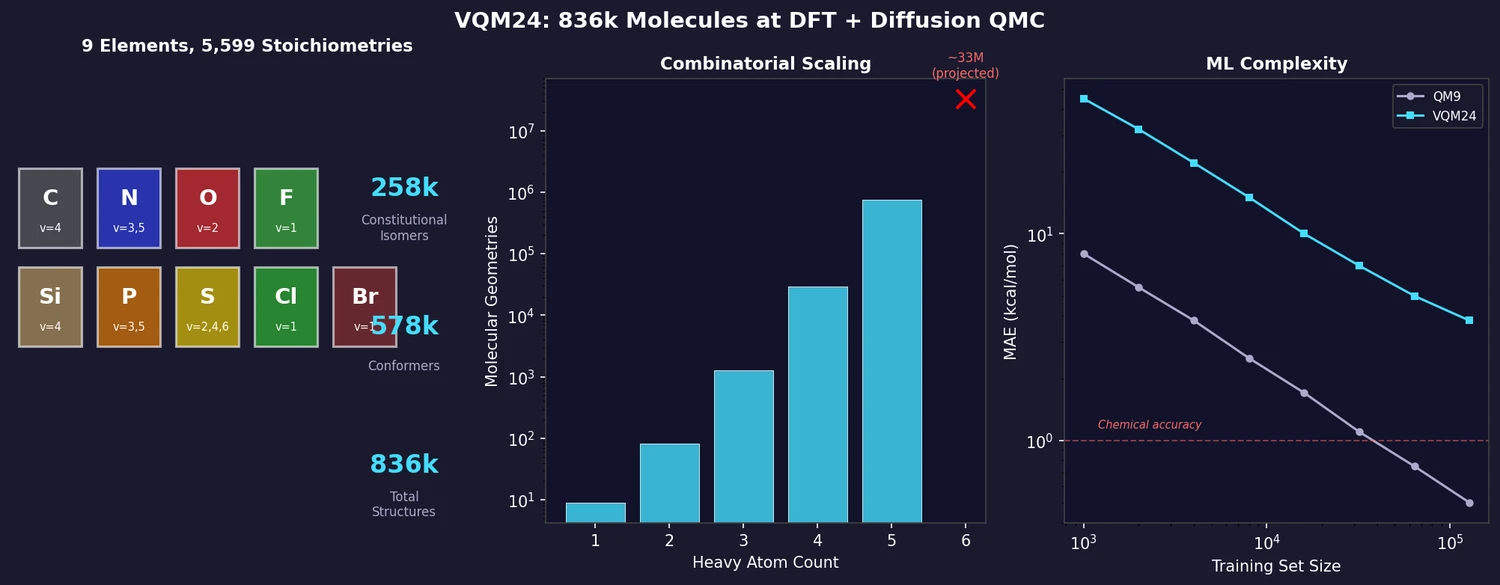
VQM24 (Vector-QM24) is the first exhaustive quantum mechanical dataset covering all possible neutral closed-shell small molecules with up to five heavy atoms from nine p-block elements (C, N, O, F, Si, P, S, Cl, Br). It provides DFT-level properties for all 836k structures and diffusion quantum Monte Carlo (DMC) energies for a 10,793-molecule subset, constituting the largest QMC dataset in chemical space to date. ML benchmarking reveals that VQM24 is significantly more challenging than QM9 despite containing smaller molecules.
Overview
Most existing QM datasets (QM7, QM9, ANI-1x) are derived from string-based molecular lists and are restricted to a few elements (typically CHONF), introducing selection bias and limiting ML model generalizability. VQM24 addresses this by exhaustively enumerating all valid stoichiometries, Lewis-rule-consistent graphs, and stable conformers for molecules composed of 9 elements with their most common valencies:
| Element | Valencies |
|---|---|
| C | 4 |
| N | 3, 5 |
| O | 2 |
| F | 1 |
| Si | 4 |
| P | 3, 5 |
| S | 2, 4, 6 |
| Cl | 1 |
| Br | 1 |
Dataset Subsets
| Heavy Atoms | Stoichiometries | Graphs | Geometries |
|---|---|---|---|
| 1 | 9 | 9 | 9 |
| 2 | 69 | 69 | 81 |
| 3 | 367 | 766 | 1,287 |
| 4 | 1,321 | 10,992 | 29,581 |
| 5 | 3,793 | 246,406 | 753,917 |
| Total | 5,559 | 258,242 | 784,875 (minima) |
Including saddle points, the full dataset contains 835,947 converged structures. Extrapolation suggests ~33 million geometries at 6 heavy atoms.
Generation Pipeline
- Stoichiometry enumeration: All combinations of up to 5 heavy atoms from the 13 element/valency types, with hydrogen counts determined by integer partitioning of total valency
- Graph generation: Constitutional isomers enumerated using Surge for each stoichiometry
- Geometry initialization: RDKit MMFF94 force field generates initial 3D coordinates
- Semi-empirical optimization: GFN2-xTB geometry optimization
- Conformer search: CREST identifies conformational isomers (~1.1M initial geometries)
- DFT optimization: Three-pass $\omega$B97X-D3/cc-pVDZ optimization in PSI4 v1.7, all using Gaussian Tight convergence criteria with density fitting (cc-pVDZ-JKFIT auxiliary basis):
- Pass 1: Default PSI4 settings (DIIS for SCF, RFO optimizer in redundant internal coordinates), max 100 steps
- Pass 2: SOSCF with full Newton step, ultrafine Lebedev-Treutler grid (590 spherical, 99 radial points), max 100 steps
- Pass 3: Full Hessian evaluation at initial geometry and every 20th step, Cartesian coordinates, max 50 steps
- DMC calculations: For 10,793 lowest-energy conformers with up to 4 heavy atoms, using QMCPACK with PBE0/ccECP/cc-pVQZ trial wavefunctions. Slater-Jastrow trial wavefunctions with Jastrow terms for 1-body (16 params/atom type, 8 Bohr cutoff), 2-body (20 params/spin-channel, 10 Bohr cutoff), and 3-body (26 params, 5 Bohr cutoff) interactions. DMC used a timestep of 0.001 a.u., 16,000 walkers, and 1,500 blocks of 40 imaginary time steps. ccECP pseudopotentials with the determinant-localization approximation and t-moves (DLTM) handled core electrons.
The $\omega$B97X-D3 functional was chosen for its strong GMTKN55 benchmark performance and for compatibility with ANI-1, ANI-1x, OrbNet Denali, QMugs, SPICE, and MultiXC-QM9, all of which use $\omega$B97X variants with double-zeta basis sets. This enables transfer learning across datasets.
Data Files and Access
The Zenodo dataset contains separate .npz files, loadable via NumPy:
| File | Contents | Molecules |
|---|---|---|
DFT_all.npz | DFT properties for all conformational minima | 784,875 |
DFT_uniques.npz | DFT properties for constitutional isomers (most stable conformer) | 258,242 |
DFT_saddles.npz | DFT properties for saddle point structures | 51,072 |
DMC.npz | DMC total energies and error bars | 10,793 |
wavefunctions.tar.gz | Wavefunction .molden files (includes MO energies) | ~106.7 GB |
All molecules are ordered consistently across every array within a file. Properties are accessed by key:
import numpy as np
data = np.load('DFT_all.npz', allow_pickle=True)
print(data.files) # list all available properties
freqs = data['freqs'] # vibrational frequencies
Computed Properties
DFT ($\omega$B97X-D3/cc-pVDZ) properties and their NPZ access keys:
| Property | Unit | Key |
|---|---|---|
| Total energies | Ha | Etot |
| Internal energies | Ha | U0 |
| Atomization energies | Ha | Eatomization |
| Electron-electron energies | Ha | Eee |
| Exchange-correlation energies | Ha | Exc |
| Dispersion energy | Ha | Edisp |
| HOMO-LUMO gap | Ha | gap |
| Dipole moments | a.u. | dipole |
| Quadrupole moments | a.u. | quadrupole |
| Octupole moments | a.u. | octupole |
| Hexadecapole moments | a.u. | hexadecapole |
| Rotational constants | MHz | rots |
| Vibrational modes | Å | vibmodes |
| Vibrational frequencies | cm$^{-1}$ | freqs |
| Gibbs free energy (H) | Ha | G |
| Internal (thermal) energy (H) | Ha | U298 |
| Enthalpy (H) | Ha | H |
| ZPVE (H) | Ha | zpves |
| Entropy (H) | cal/mol K | S |
| Heat capacities (H) | cal/mol K | Cv, Cp |
| Electrostatic potentials at nuclei | a.u. | Vesp |
| Mulliken charges | a.u. | Qmulliken |
| SMILES | graphs | |
| InChI strings | inchi |
(H) indicates thermodynamic properties computed via the harmonic approximation. Molecular orbital energies are available in the wavefunction .molden files.
DMC properties (DMC.npz) include total energy (Etot) and statistical error bar (std) for each molecule.
DMC energies (PBE0/ccECP/cc-pVQZ nodal surfaces, Slater-Jastrow trial wavefunctions) achieve average statistical uncertainty of 0.4 mHa across ~2.3 billion samples per molecule.
ML Benchmarking: Harder Than QM9
Learning curves for atomization energy prediction show that VQM24 is substantially more challenging than QM9 for all tested models:
- KRR models (CM, ACSF, LMBTR, FCHL19, cMBDF) and GNNs (SchNet, PaiNN) all show up to ~8x larger mean errors on VQM24 than QM9 at the same training set size
- None of the tested models achieve chemical accuracy (1 kcal/mol) on VQM24, even with 128k training molecules
- The atomization energy range in VQM24 (1,545 kcal/mol) is smaller than QM9 (2,427 kcal/mol), so the higher errors reflect greater chemical diversity rather than a wider property range
- For a fair comparison with QM9 (which has no conformational isomers), learning curves use only the 258k unique constitutional isomers from VQM24
Benchmark methodology: KRR models use an atomic Gaussian kernel with hyperparameters (length-scale $l$, regularizer $\lambda$) optimized via grid search and 5-fold cross-validation. Both GNNs (SchNet, PaiNN) use 128 atomic basis functions (589k total parameters), trained for 1,000 epochs with Adam (lr = $10^{-4}$). Test set size is 10,000 randomly selected molecules, with results averaged over 5 runs. Training and evaluation scripts are available in the GitHub repository.
Prediction error analysis with the best KRR model (cMBDF, trained on 200k across 4 disjoint training sets on all 784,875 equilibrium geometries) yields an overall MAE of 0.75 kcal/mol (standard deviation 1.55 kcal/mol). The largest individual error reaches 167.3 kcal/mol, and the 25 largest outliers have a mean absolute error of 85.9 kcal/mol.
Strengths & Limitations
Strengths:
- Exhaustive coverage of 1-5 heavy atom chemical space across 9 elements
- Both DFT and DMC-level data (largest QMC dataset in chemical space)
- Includes conformational isomers (average 3 per constitutional isomer)
- Extensive property set including wavefunctions and multipole moments up to hexadecapole
- More challenging ML benchmark than QM9, exposing model limitations
Limitations:
- Limited to 5 heavy atoms (very small molecules)
- 262,542 structures (~24%) failed DFT convergence, with a strong silicon bias in failures
- 51,072 structures converged to saddle points rather than minima
- DMC subset limited to 4 heavy atoms (10,793 molecules)
- Does not include metals, rare gases, or heavier halogens (I)
Reproducibility Details
Status: Highly Reproducible
The paper, dataset, and code are all publicly available.
| Artifact | Type | License | Notes |
|---|---|---|---|
| VQM24 Dataset (Zenodo) | Dataset | CC-BY-4.0 | DFT .npz files + DMC .npz + wavefunction tarball (~108 GB total) |
| dkhan42/VQM24 (GitHub) | Code | MIT | Generation tools, PSI4 templates, KRR and GNN training scripts |
| arXiv preprint | Paper | arXiv license | Open-access preprint of the Scientific Data article |
Software stack: Surge (graph enumeration), RDKit/MMFF94 (initial geometries), GFN2-xTB (semi-empirical optimization), CREST (conformer search), PSI4 v1.7 (DFT), PySCF (trial wavefunctions), QMCPACK (DMC), QMLcode (KRR models), SchNetPack (GNN models).
Hardware requirements:
- DFT: Three-pass $\omega$B97X-D3/cc-pVDZ optimization in PSI4 (compute details not specified per-molecule for DFT)
- DMC trial wavefunctions: Argonne LCRC Improv, single node (2x AMD EPYC 7713, 64 cores, 2 GHz), ~45 seconds per molecule, ~134 node-hours total
- DMC calculations: Argonne Polaris HPC (AMD EPYC 7543P, 64 cores, 2.8 GHz), 20 nodes per molecule, ~15 minutes each, ~54,000 node-hours total
Citation
@article{khan2025quantum,
title={Quantum mechanical dataset of 836k neutral closed-shell molecules
with up to 5 heavy atoms from C, N, O, F, Si, P, S, Cl, Br},
author={Khan, Danish and Benali, Anouar and Kim, Scott Y. H.
and von Rudorff, Guido Falk and von Lilienfeld, O. Anatole},
journal={Scientific Data},
volume={12},
number={1},
pages={1551},
year={2025},
publisher={Nature Portfolio},
doi={10.1038/s41597-025-05428-4}
}