Key Contribution
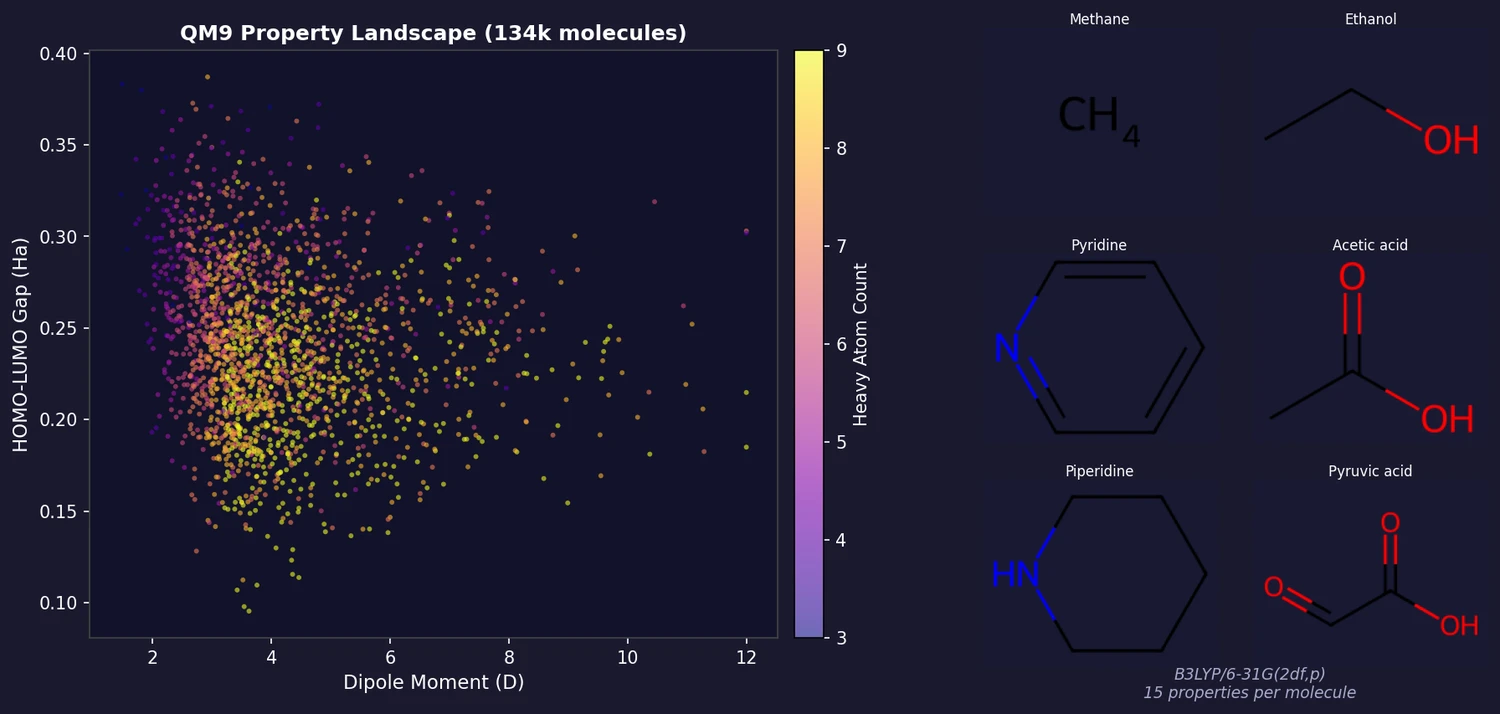
QM9 provides a consistent, comprehensive set of quantum chemical properties for 133,885 small organic molecules (up to 9 heavy atoms of C, N, O, F) from the GDB-17 chemical universe. It is among the most widely used benchmark datasets in molecular machine learning, enabling systematic development and evaluation of structure-property prediction methods.
Overview
The dataset corresponds to the GDB-9 subset of the GDB-17 chemical universe: all neutral molecules with up to nine heavy atoms (C, O, N, F), not counting hydrogen. Cations, anions, and molecules containing S, Br, Cl, or I were excluded, though 1,705 zwitterions (relevant for small biomolecules like amino acids) were retained. The dataset spans 621 stoichiometries. It includes small amino acids (glycine, alanine), nucleobases (cytosine, uracil, thymine), and pharmaceutically relevant building blocks (pyruvic acid, piperazine, hydroxy urea).
Computed Properties
All properties were calculated at the B3LYP/6-31G(2df,p) level of DFT. The 15 scalar properties per molecule are:
| Property | Unit | Description |
|---|---|---|
| A, B, C | GHz | Rotational constants |
| $\mu$ | D | Dipole moment |
| $\alpha$ | $a_0^3$ | Isotropic polarizability |
| $\varepsilon_{\text{HOMO}}$ | Ha | HOMO energy |
| $\varepsilon_{\text{LUMO}}$ | Ha | LUMO energy |
| $\varepsilon_{\text{gap}}$ | Ha | HOMO-LUMO gap |
| $\langle R^2 \rangle$ | $a_0^2$ | Electronic spatial extent |
| ZPVE | Ha | Zero-point vibrational energy |
| $U_0$ | Ha | Internal energy at 0 K |
| $U$ | Ha | Internal energy at 298.15 K |
| $H$ | Ha | Enthalpy at 298.15 K |
| $G$ | Ha | Free energy at 298.15 K |
| $C_v$ | cal/mol K | Heat capacity at 298.15 K |
Each molecule is stored in an extended XYZ file. The first line gives the atom count, and the second (comment) line packs all 15 scalar properties. Lines 3 through $n_a + 2$ contain element type, Cartesian coordinates (x, y, z in Angstroms), and Mulliken partial charges as a fifth column. Three trailing lines append harmonic vibrational frequencies ($3n_a - 5$ or $3n_a - 6$ modes, in cm$^{-1}$), SMILES strings (from GDB-17 and from the B3LYP-relaxed geometry), and InChI strings (from Corina and B3LYP geometries).
Dataset Subsets
| Subset | Size | Description |
|---|---|---|
| GDB-9 (Full) | 133,885 | All molecules, B3LYP properties |
| C7H10O2 isomers | 6,095 | Predominant stoichiometry, with additional G4MP2 energetics |
| Validation set | 100 | Random subset with G4MP2, G4, and CBS-QB3 reference values |
Geometry Generation Pipeline
Starting from GDB-17 SMILES strings, initial 3D coordinates were generated with Corina, then relaxed at the PM7 semi-empirical level (MOPAC), followed by B3LYP/6-31G(2df,p) geometry optimization (Gaussian 09). A five-stage iterative convergence procedure handled difficult cases: default thresholds, then ultrafine grids, tighter SCF criteria, Hessian-guided optimization (calcfc), and full Hessian optimization (calcall). After all stages, 11 molecules still failed to converge to true minima (6 converged with loose thresholds, 2 near-linear molecules converged to saddle points with very low imaginary frequencies below $i10 \text{ cm}^{-1}$).
Validation
Geometry consistency: B3LYP-relaxed geometries were converted back to InChI strings and compared against the original GDB-17 InChI. 3,054 molecules failed this round-trip test, primarily due to implementation-specific artifacts in SMILES/InChI conversion rather than actual geometry problems. Coulomb-matrix distances between Corina and B3LYP geometries quantified the magnitude of geometric changes.
Energy accuracy: For 100 randomly selected molecules, B3LYP atomization enthalpies were compared against higher-level composite methods. These reference methods are themselves near experimental accuracy: G4MP2 achieves MAE 1.0 and RMSE 1.5 kcal/mol against the G3/05 test set of 454 experimental energies, while G4 achieves MAE 0.8 and RMSE 1.2 kcal/mol on the same set. G4MP2 also deviates by only 1.4 kcal/mol from the highly accurate W1w composite procedure on 261 bond dissociation enthalpies (BDE261 dataset). Against these references, B3LYP shows:
| Reference | MAE (kcal/mol) | RMSE (kcal/mol) | Max AE (kcal/mol) |
|---|---|---|---|
| G4MP2 | 5.0 | 6.1 | 16.0 |
| G4 | 4.9 | 5.9 | 14.4 |
| CBS-QB3 | 4.5 | 5.5 | 13.4 |
All 6,095 C7H10O2 isomers passed the geometry consistency check, and their G4MP2-level energetics provide a higher-accuracy benchmark within a fixed stoichiometry.
Strengths & Limitations
Strengths:
- Comprehensive and consistent: same level of theory across all 134k molecules
- Derived from a systematically enumerated chemical space (GDB-17), reducing selection bias
- Rich property set covering geometric, electronic, energetic, and thermodynamic quantities
- Widely adopted benchmark enabling reproducible comparisons across ML methods
Limitations:
- Restricted to very small molecules (up to 9 heavy atoms), limiting relevance to drug-sized compounds
- Only CHONF elements, excluding sulfur, halogens (Cl, Br, I), and metals
- B3LYP/6-31G(2df,p) has known systematic errors (~5 kcal/mol MAE for atomization enthalpies)
- 3,054 molecules have geometry consistency issues in SMILES/InChI round-tripping
- Single conformer per molecule (energy-minimized geometry only)
Reproducibility Details
| Artifact | Type | License | Notes |
|---|---|---|---|
| Figshare collection | Dataset | CC BY-NC-SA 4.0 | Full dataset: 134k molecules, C7H10O2 isomers, validation set, atomic references |
The Figshare deposit contains four files:
dsgdb9nsd.xyz.tar.bz2: All 133,885 GDB-1 through GDB-9 molecules with B3LYP propertiesdsC7O2H10nsd.xyz.tar.bz2: 6,095 C7H10O2 constitutional isomers with G4MP2 energeticsvalidation.txt: Atomization enthalpies at B3LYP, G4MP2, G4, and CBS-QB3 for 100 random moleculesatomref.txt: Atomic reference energies for computing atomization energies from total energies
All data is in extended XYZ plain-text format. The paper and its metadata are open access (CC BY-NC-SA 4.0 for the article, CC0 for metadata).
No source code is provided. The computational pipeline relies on commercial and semi-commercial software: Corina (3D coordinate generation), MOPAC (PM7 semi-empirical relaxation), and Gaussian 09 (B3LYP DFT calculations). Specific convergence keywords and iteration procedures are documented in the paper. Hardware requirements are not reported.
Reproducibility status: Partially Reproducible. The dataset itself is fully available, but regenerating it requires commercial licenses for Corina and Gaussian 09.
Citation
@article{ramakrishnan2014quantum,
title={Quantum chemistry structures and properties of 134 kilo molecules},
author={Ramakrishnan, Raghunathan and Dral, Pavlo O. and Rupp, Matthias and von Lilienfeld, O. Anatole},
journal={Scientific Data},
volume={1},
number={1},
pages={140022},
year={2014},
publisher={Nature Portfolio},
doi={10.1038/sdata.2014.22}
}