A Graph-Grammar and ILP Framework for Pathway Discovery
Abel et al. present a Method paper that couples generative chemical space expansion with integer linear programming (ILP) pathway queries to systematically propose artificial carbon fixation pathways. The workflow uses the cheminformatics package MØD to iteratively expand a reaction hypergraph from a seed set of metabolites and rule-based enzyme reactions, then queries the resulting network for autocatalytic flows producing a chosen target molecule. Post-hoc annotation with eQuilibrator Gibbs energies and cofactor accounting ranks candidates by thermodynamic feasibility. Applied to the Acetyl-CoA-Succinyl-CoA pathway family plus selected synthetic and theoretical pathways, the framework recovers the natural pathways and proposes two new theoretical autocatalytic cycles (an 11-step Acetyl-CoA cycle and a 12-step Malate cycle) whose efficiency, measured in ATP and redox cofactors per fixed carbon, is comparable to the synthetic CETCH cycle and the natural rTCA.
Why Computational Pathway Design for Carbon Fixation
Fixing atmospheric CO$_2$ or bicarbonate into value-added chemicals is a thermodynamically unfavorable process that nature solves through enzymatic cascades coupled to cofactor-driven reactions. Seven natural carbon fixation pathways are known, along with several artificial proposals, and the Acetyl-CoA-Succinyl-CoA family is particularly appealing as a design template because each member overlaps structurally with at least one other and each exhibits autocatalysis. Prior approaches to artificial pathway design (e.g., Erb Lab CETCH, HOPAC) rely heavily on manual heuristics, database searches, and extensive in-vitro optimization including directed evolution. Earlier computational work (Löwe and Kremling, 2021) uses flux balance analysis and expert curation that requires complete kinetic parameterization, making generative exploration infeasible. Abel et al. target the design stage directly: a computational approach that can quickly enumerate many topologically distinct pathway candidates without requiring a priori kinetic parameters.
Generative Chemical Space Expansion with Graph-Grammar Rules
The core innovation is treating the chemical reaction network (CRN) as a directed multi-hypergraph $H = (V, E)$ where vertices in $V$ are molecules and each hyperedge $e \in E$ is a directed pair $(e_{tail}, e_{head})$ of multisets representing reactants and products. This hyperedge formalization directly captures the many-to-many nature of biochemical reactions.
Reactions are specified as graph transformation rules written in the Graph Modeling Language (GML). A rule defines the bond rewiring at a reaction center plus a tunable molecular context around that center. A rule with no context is fully promiscuous (every oxidoreductase class reaction, say); a rule with rich context mimics a specific enzyme. This rule-based formalism lets one rule represent an entire reaction class, so the CRN can be expanded without enumerating every possible enzyme-substrate pair in advance. Expansion proceeds iteratively: the rules act on the current molecule pool, producing new molecules and new hyperedges, until a user-defined step count is reached. Two biochemical sanity constraints bound the combinatorial explosion: molecules are restricted to at most 6 carbon atoms in the backbone (excluding the CoA moiety), and at most one CoA group per molecule.
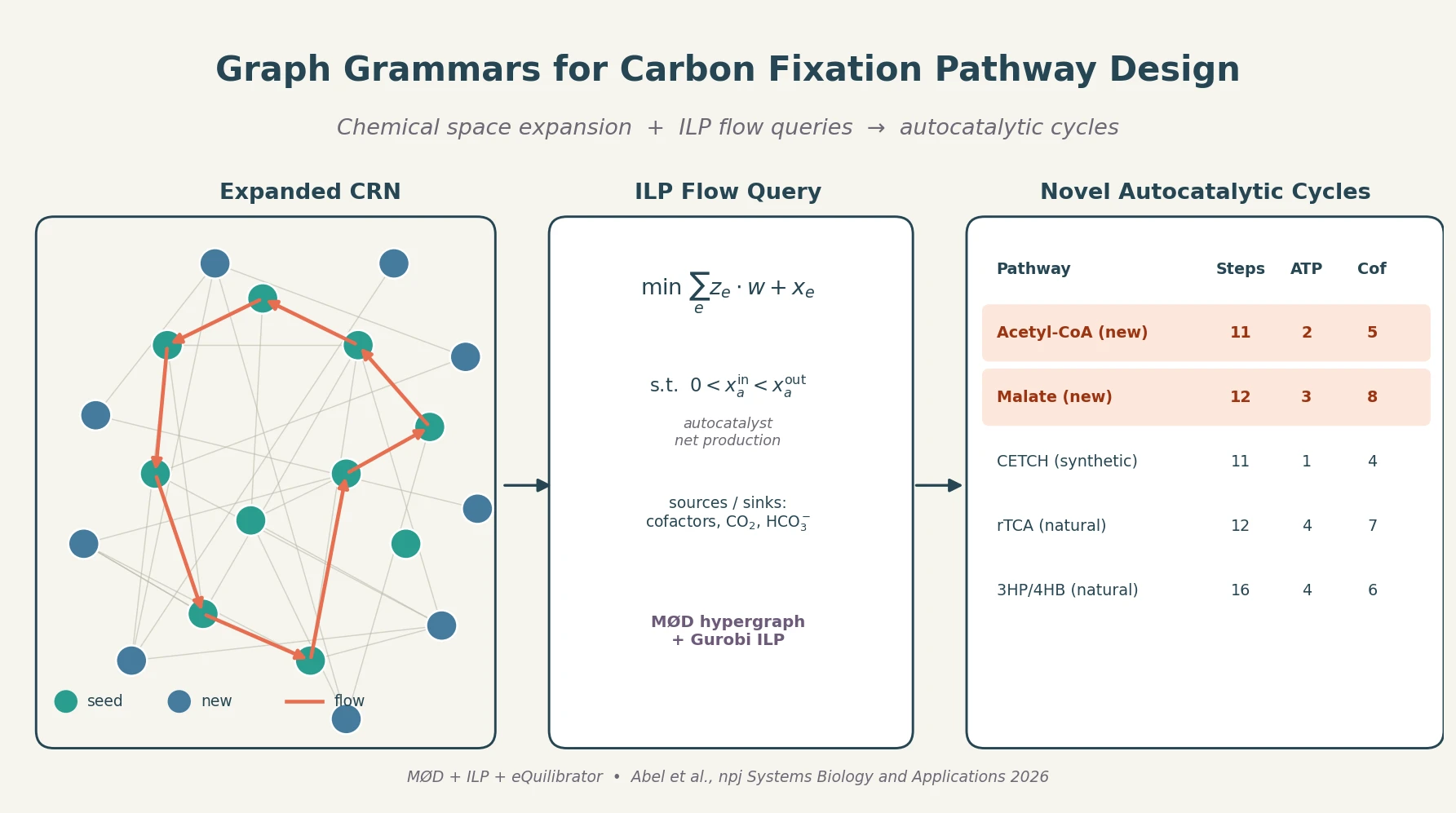
Pathway discovery is then an ILP flow query over the CRN. A pathway is a hyperflow: an assignment of integer flow values to hyperedges such that internal molecules balance between production and consumption, leaving only designated source and sink molecules with net flow. The main optimization objective minimizes the number of reactions used and, as a tiebreaker, the magnitude of flow on those reactions:
$$ \min \left(\sum_{e \in E} z_e \cdot w + x_e\right) $$
where $z_e$ is a boolean indicator that hyperedge $e$ carries flow, $x_e$ is the integer flow on $e$, and the weight $w = 1000$ prioritizes minimizing the edge count over the total flow magnitude. Autocatalysis is encoded as a constraint on the autocatalyst molecule $a$: its inflow and outflow are both positive, with outflow strictly exceeding inflow so the cycle nets at least one additional molecule of the autocatalyst.
$$ 0 < x_a^{in} < x_a^{out} $$
Only the autocatalyst itself, cofactors, and CO$_2$/HCO$_3^-$ are permitted as sources and sinks, so any valid flow represents a net reaction that fixes carbon and regenerates the autocatalyst. Unlike classical flux balance analysis, which optimizes continuous flux distributions at steady state, the integer-valued ILP formulation emphasizes pathway structure (which reactions are active) rather than flux magnitude.
Solutions are post-annotated with two feasibility measures. The first is cofactor accounting, split into ATP/ADP as an energy proxy and reduced redox cofactors (NAD(P)H, ubiquinone, Ferredoxin) as an electron proxy. The second is the standard Gibbs free energy of the net reaction computed via the eQuilibrator 3.0 component-contribution method at pH 7 and ionic strength 0.1 M using the eQuilibrator API 0.6.0:
$$ \Delta_r G’^{\circ} = \sum \Delta_f G’^{\circ}_{\text{products}} - \sum \Delta_f G’^{\circ}_{\text{reactants}} $$
Experimental Setup, Queries, and Comparison to Literature
The seed pool for expansion contains 49 intermediates drawn from the Acetyl-CoA-Succinyl-CoA family (rTCA, DC/4-HB, 3-HP/4-HB, 3-HP bicycle), the synthetic CETCH cycle, and theoretical pathways proposed by Bar-Even et al., plus 20 helper molecules (cofactors, water, CO$_2$). Rule contexts were derived from KEGG enzyme entries. The Calvin-Benson-Basham cycle and the non-autocatalytic Wood-Ljungdahl and reductive glycine pathways were excluded.
Expansion statistics (Table 4 in the paper):
| Expansion steps | Molecules (vertices) | Reactions (hyperedges) |
|---|---|---|
| 1 | 165 | 220 |
| 2 | 318 | 942 |
| 5 | 996 | 29,266 |
At one expansion step, flow queries recover only the input pathways with no recombinations. Two expansion steps produce sufficient novelty for recombined pathways while keeping ILP runtimes tractable. Five steps makes flow queries computationally prohibitive without adding biological insight. All reported analyses use the two-step CRN.
Three benchmark flow queries target autocatalytic pathways producing Acetyl-CoA, Malate, and Propionyl-CoA. Each query is run to return 1000 topologically distinct optimal solutions (under the ILP objective, solutions with equal length are equally optimal). All flow queries were solved with Gurobi 11.0.3 under an academic license on a consumer laptop (AMD Ryzen 7 5700U, 16 GB RAM, Windows 11). The full 1000-solution search took just under 18 hours.
Two Novel Autocatalytic Cycles Competitive with Synthetic Pathways
The shortest-pathway queries yield two novel theoretical autocatalytic cycles: an 11-step Acetyl-CoA cycle and a 12-step Malate cycle. Comparison to natural, theoretical, and synthetic pathways on the four standard measures (steps, ATP units, cofactors, carbon units fixed per cycle):
| Pathway | Status | Steps | ATP | Cofactors | C fixed | ATP/C | Cof/C |
|---|---|---|---|---|---|---|---|
| Shortest Acetyl-CoA (this work) | Theoretical | 11 | 2 | 5 | 2 | 1 | 2.5 |
| Shortest Malate (this work) | Theoretical | 12 | 3 | 8 | 4 | 0.75 | 2 |
| CETCH | Synthetic | 11 | 1 | 4 | 2 | 0.5 | 2 |
| rGPS-MCG | Synthetic | 18 | 4 | 6 | 3 | 1.33 | 2 |
| C4-glyoxylate / alanine | Theoretical | 9 | 2 | 2 | 2 | 1 | 1 |
| rTCA | Natural | 12 | 4 | 7 | 4 | 1 | 1.75 |
| 3HP/4HB | Natural | 16 | 4 | 6 | 2 | 2 | 3 |
| DC/4HB | Natural | 14 | 4 | 7 | 2 | 2 | 3.5 |
| 3HP-bicycle | Natural | 19 | 3 | 4 | 2 | 1.5 | 2 |
The 11-step Acetyl-CoA cycle matches CETCH in length and carbon units fixed while using one more ATP and one more redox cofactor. The Malate cycle is the same length as rTCA (12 steps) but uses one fewer ATP and one fewer cofactor while fixing the same four carbons.
Across the 1000-solution benchmarks (Table 2 of the paper), the Acetyl-CoA cycle is the most cofactor-efficient per step (0.69 cofactors/step; average 7.6 total), while Propionyl-CoA and Malate average 0.89 and 0.88 cofactors/step. Gibbs energies average $\Delta_r G’^{\circ} = -150.66$ kJ/mol for Acetyl-CoA, $-165.82$ for Propionyl-CoA, and $-196.98$ for Malate, making the Malate query the most thermodynamically driven even after accounting for its higher cofactor count. Three specific Acetyl-CoA solutions inspected in detail share a common rTCA-like core with a glyoxylate shunt and differ mainly along the oxaloacetate-to-malyl-CoA branch; their totals range from $\Delta_r G’^{\circ}_{total} = -80$ kJ/mol (the one-ATP solution) to $-168$ kJ/mol.
All solutions rely on Ferredoxin-dependent carboxylating enzymes (pyruvate:ferredoxin oxidoreductase and 2-ketoglutarate:ferredoxin oxidoreductase), which have higher reduction potentials than NAD(P) but are oxygen-sensitive and would restrict wet-lab implementation to anaerobic conditions or engineered anaerobic strains.
Findings, Limitations, and Future Directions
The workflow produces pathway candidates whose efficiency approaches the best synthetic designs while running on a consumer laptop, and it generalizes to any chemical space that can be formalized by graph-transformation rules. Because the ILP returns many equally optimal solutions, a downstream filtering step can select candidates matching user criteria (oxygen sensitivity, specific cofactor preference, enzyme availability).
Acknowledged limitations include: the topology-only search ignores enzyme kinetics, so candidates that look thermodynamically favorable might be bottlenecked in practice; the carbon-count and CoA restrictions are necessary to bound combinatorial blow-up but also constrain the discoverable space; reliance on Ferredoxin complicates implementation; and enzyme availability varies across organisms, which matters for recombination-based designs. The authors point to kinetic modeling, cofactor-recycling system inclusion, and incorporation of metabolic reactions outside the canonical carbon fixation space as future directions.
The paper positions itself as a design-stage tool rather than an end-to-end in-vitro pipeline. The authors frame the contribution as idea generation that complements, not replaces, the subsequent experimental optimization (enzyme engineering, directed evolution) that has carried prior synthetic pathway work from theory to in-vitro success.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Seed molecules | Curated Acetyl-CoA-Succinyl-CoA family + CETCH + Bar-Even theoretical | 49 metabolites + 20 cofactors | Tables S1-S2 |
| Reaction rules | KEGG enzyme entries, GML-encoded | Rules listed in Figure S1 | Conservative context |
| CRN (2-step expansion) | Generated by MØD | 318 molecules, 942 reactions | Primary analysis space |
| Thermodynamic data | eQuilibrator 3.0 component-contribution | All molecules in space | pH 7, ionic strength 0.1 M |
Algorithms
Graph-grammar rule expansion via MØD 1.0.0 with a 6-carbon backbone cap and at most one CoA moiety per molecule. ILP flow queries formulated with the edge-minimization objective in Equation (1) and the autocatalysis constraint in Equation (2). Natural pathway presence first verified via set operations on the CRN, then reconfirmed by constraining the ILP to pass through core intermediates. The pathway solution enumeration is structural: 1000 topologically distinct solutions per query at the optimal objective value.
Models
No machine-learning models. The pipeline is symbolic: graph transformations, hypergraph flow constraints, and component-contribution free energy estimates.
Evaluation
| Metric | Acetyl-CoA | Propionyl-CoA | Malate |
|---|---|---|---|
| Avg steps | 11 | 15 | 12 |
| Avg cofactors | 7.6 | 13.3 | 10.6 |
| Cofactors/step | 0.69 | 0.89 | 0.88 |
| Avg $\Delta_r G’^{\circ}$ (kJ/mol) | $-150.66$ | $-165.82$ | $-196.98$ |
Hardware
Gurobi 11.0.3 (academic license) on a consumer laptop: AMD Ryzen 7 5700U, 16 GB RAM, Windows 11. Full 1000-solution runs for the three benchmark queries completed in just under 18 hours total.
Artifacts and licensing
- Code and output pathways: github.com/anne-susann/C_fixation_pathway_design (MIT License)
- MØD cheminformatics package (version 1.0.0)
- eQuilibrator API version 0.6.0
- Gurobi 11.0.3
Paper Information
Citation: Abel, A.-S., Lauber, N., Andersen, J. L., Fagerberg, R., Merkle, D. E., & Flamm, C. (2026). Computational approaches in chemical space exploration for carbon fixation pathways. npj Systems Biology and Applications, 12(1), 17. https://doi.org/10.1038/s41540-025-00641-8
Publication: npj Systems Biology and Applications, 2026
@article{abel2026computational,
title={Computational approaches in chemical space exploration for carbon fixation pathways},
author={Abel, Anne-Susann and Lauber, Nino and Andersen, Jakob Lykke and Fagerberg, Rolf and Merkle, Daniel Elmar and Flamm, Christoph},
journal={npj Systems Biology and Applications},
volume={12},
number={1},
pages={17--17},
year={2026},
publisher={Nature Portfolio},
doi={10.1038/s41540-025-00641-8}
}