MolPMoFiT: Inductive Transfer Learning for QSAR
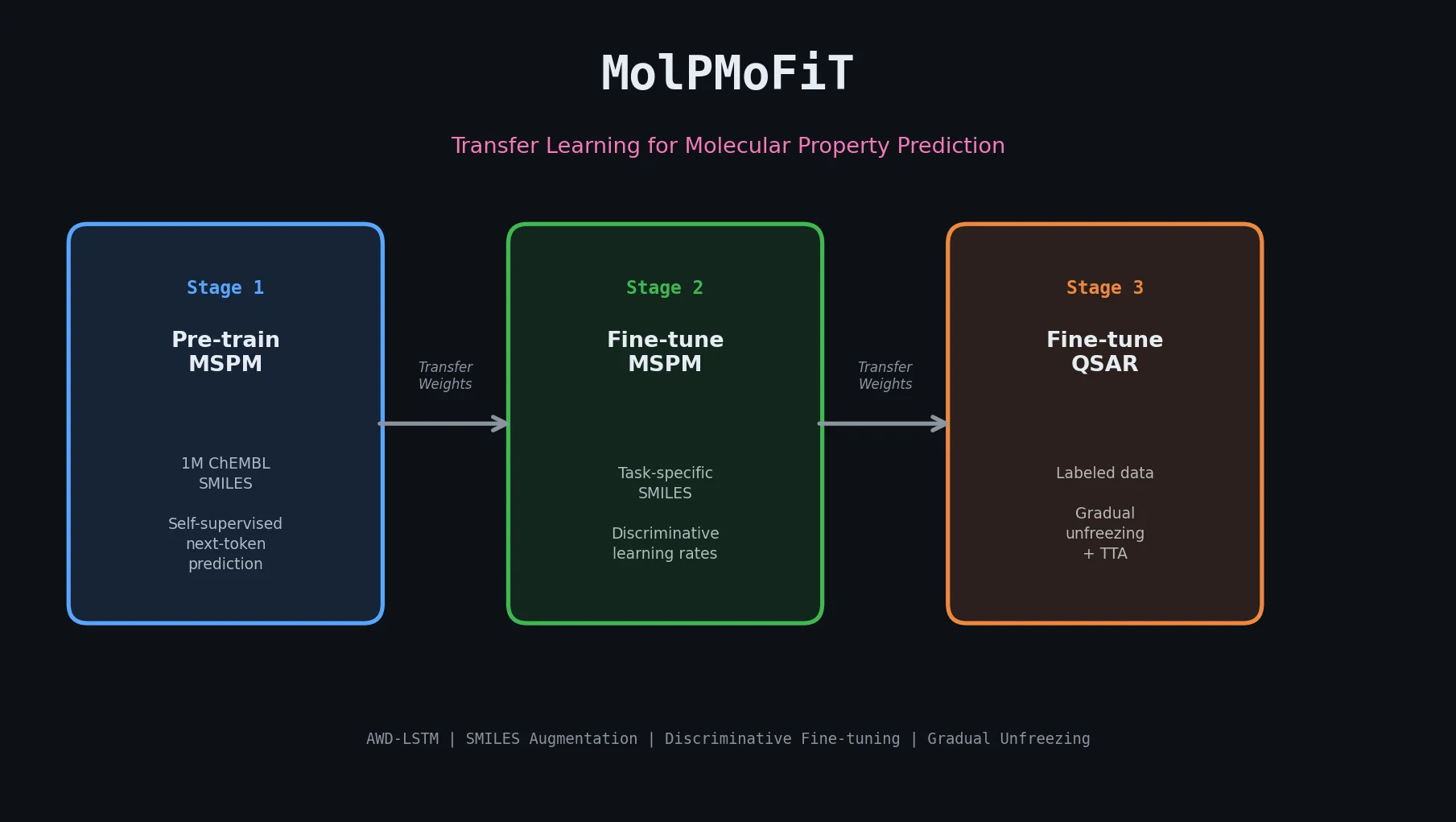
MolPMoFiT applies ULMFiT-style transfer learning to QSAR modeling, pre-training an AWD-LSTM on one million ChEMBL molecules and fine-tuning for property prediction on small datasets.
MolPMoFiT applies ULMFiT-style transfer learning to QSAR modeling, pre-training an AWD-LSTM on one million ChEMBL molecules and fine-tuning for property prediction on small datasets.
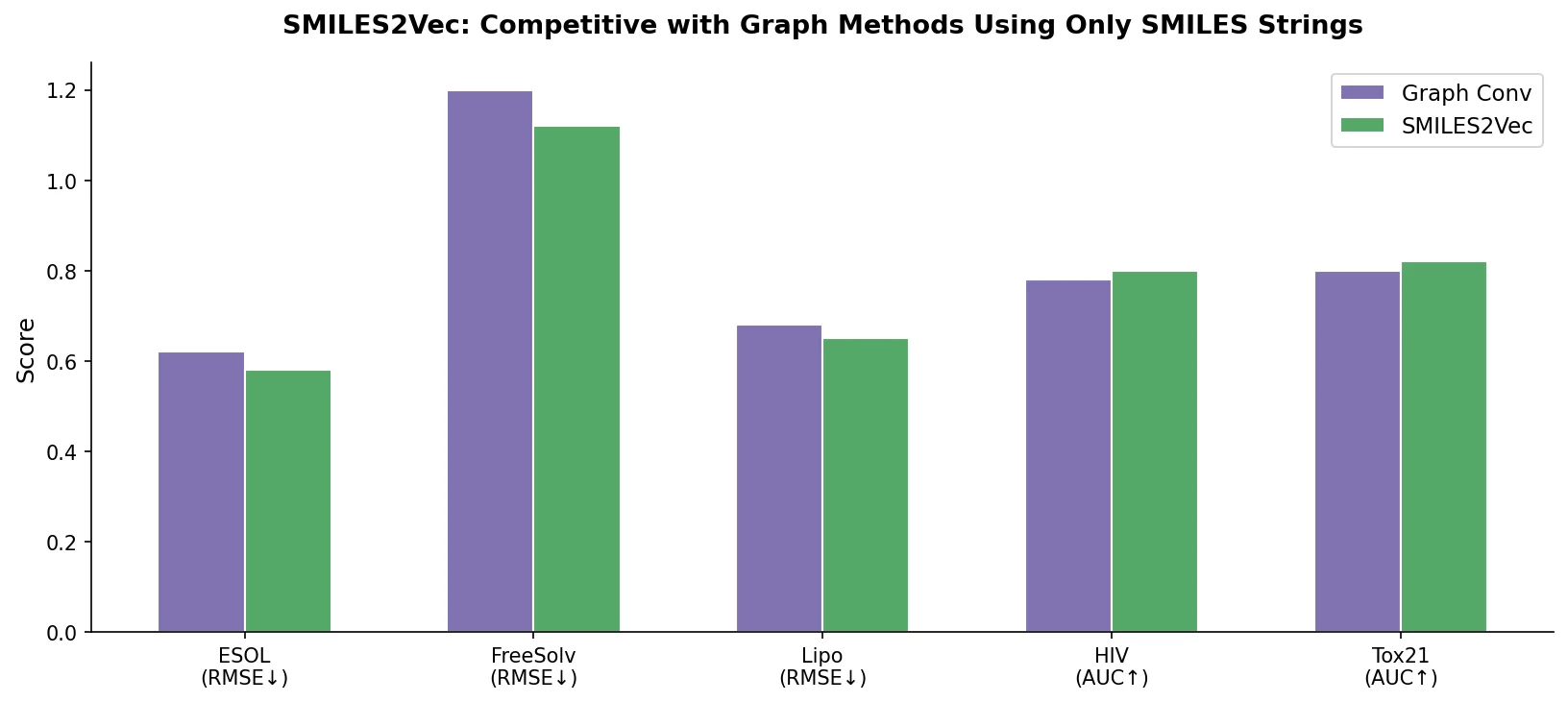
SMILES2Vec is a deep RNN that learns chemical features directly from SMILES strings using a Bayesian-optimized CNN-GRU architecture. It matches graph convolution baselines on toxicity and activity prediction, and its explanation mask identifies chemically meaningful functional groups with 88% accuracy.
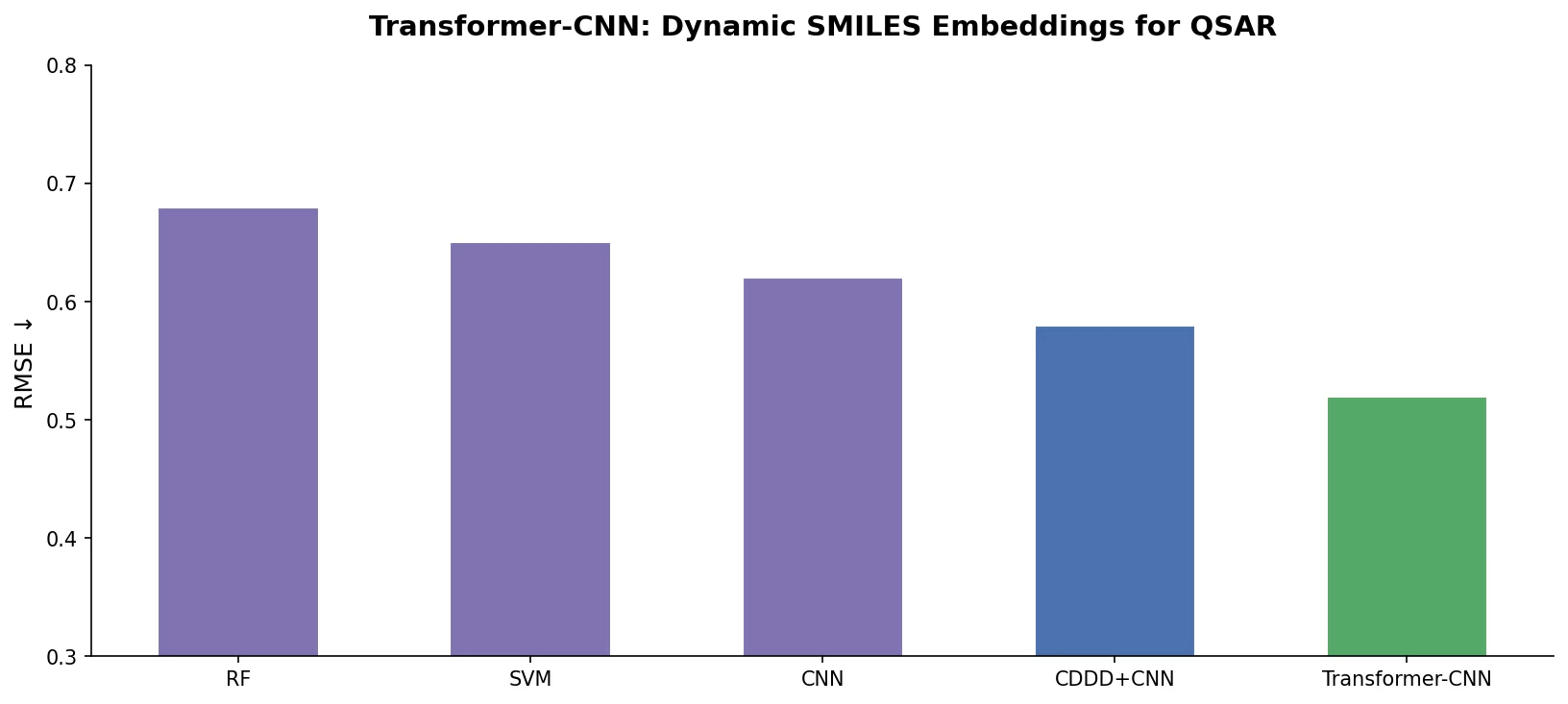
Transformer-CNN extracts dynamic SMILES embeddings from a Transformer trained on SMILES canonicalization and feeds them to a TextCNN for QSAR modeling, achieving strong results across 18 benchmarks with built-in LRP interpretability.
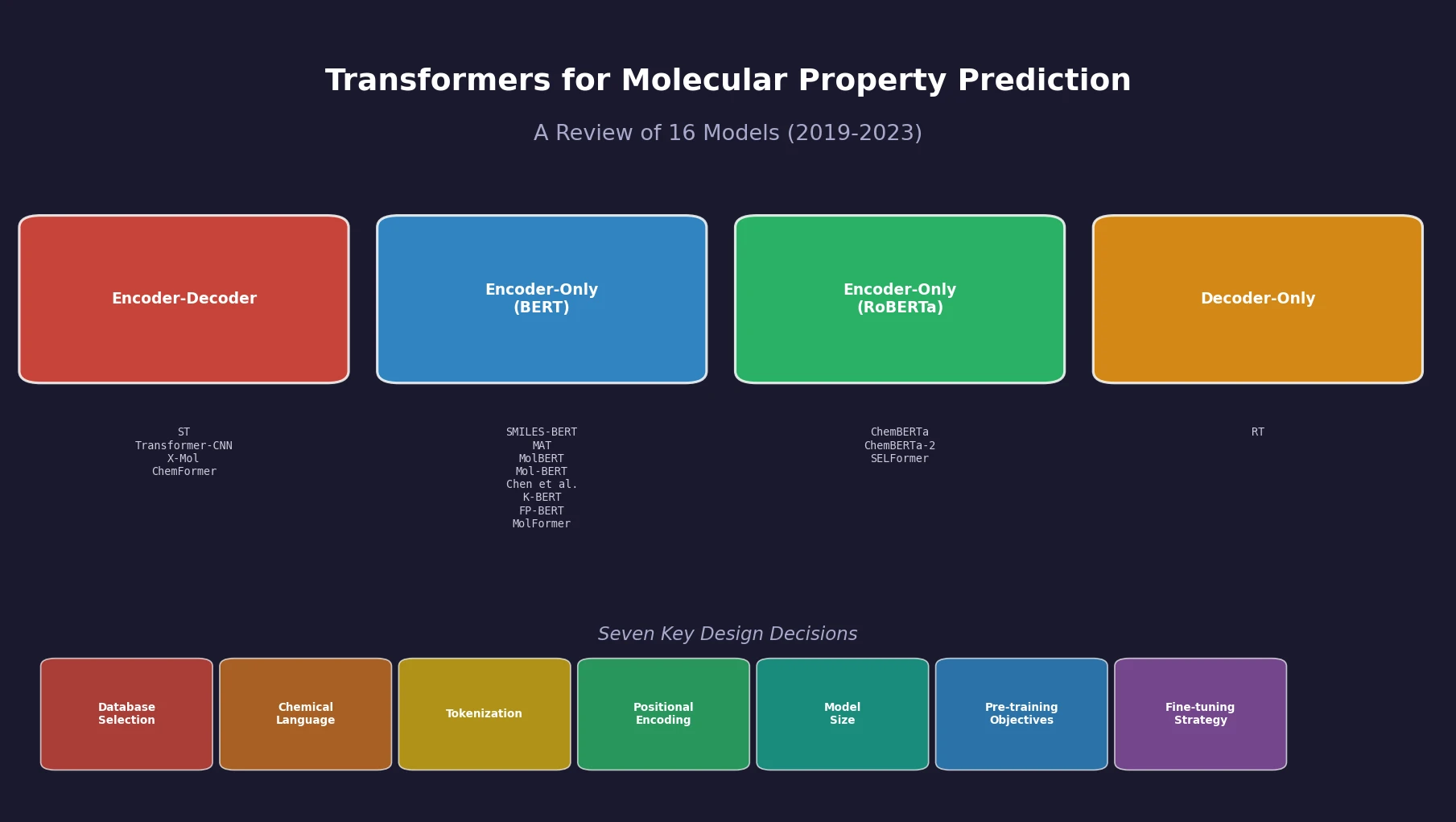
Sultan et al. review 16 sequence-based transformer models for molecular property prediction, systematically analyzing seven design decisions (database selection, chemical language, tokenization, positional encoding, model size, pre-training objectives, and fine-tuning strategy) and identifying a critical need for standardized evaluation practices.
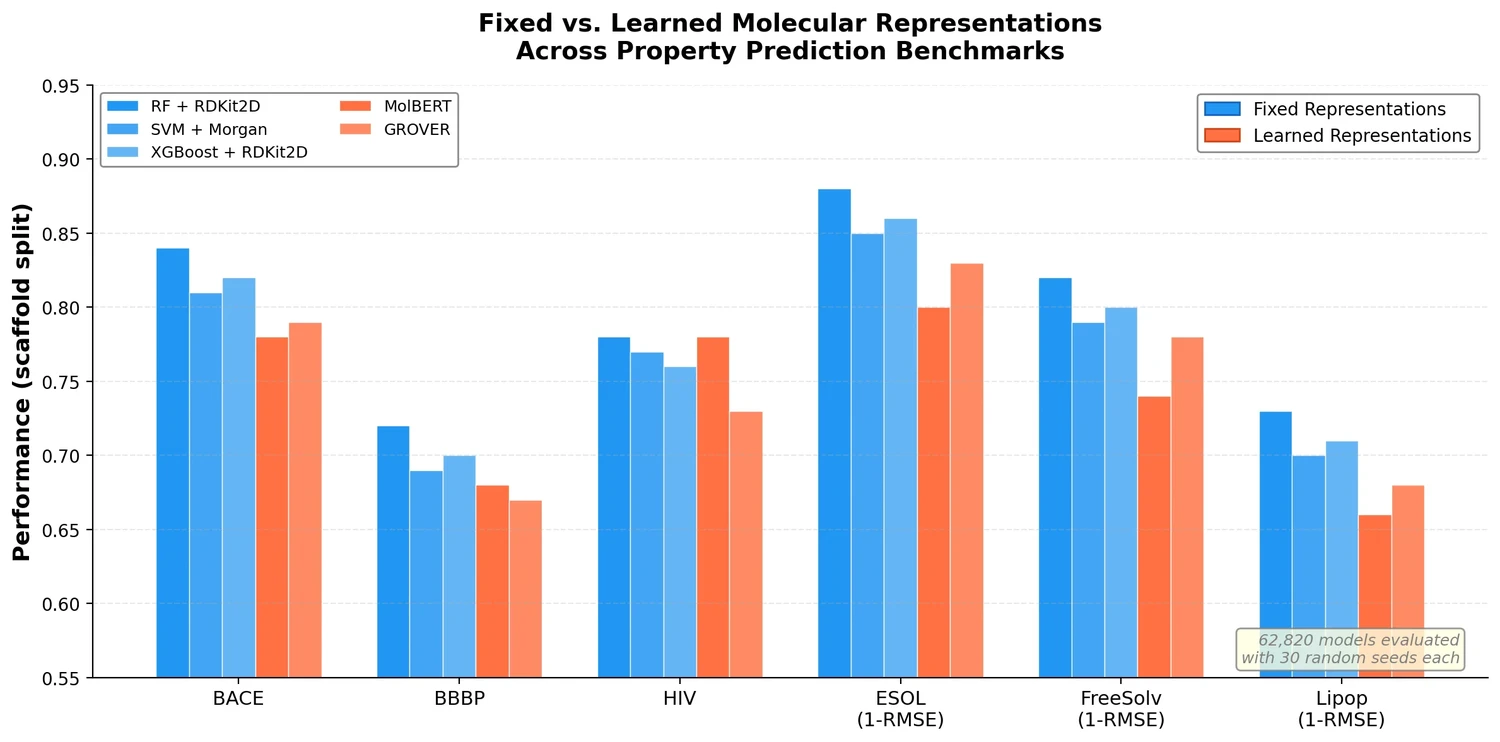
This study trains over 62,000 models to systematically evaluate molecular representations and models for property prediction, finding that traditional ML on fixed descriptors often outperforms deep learning approaches.
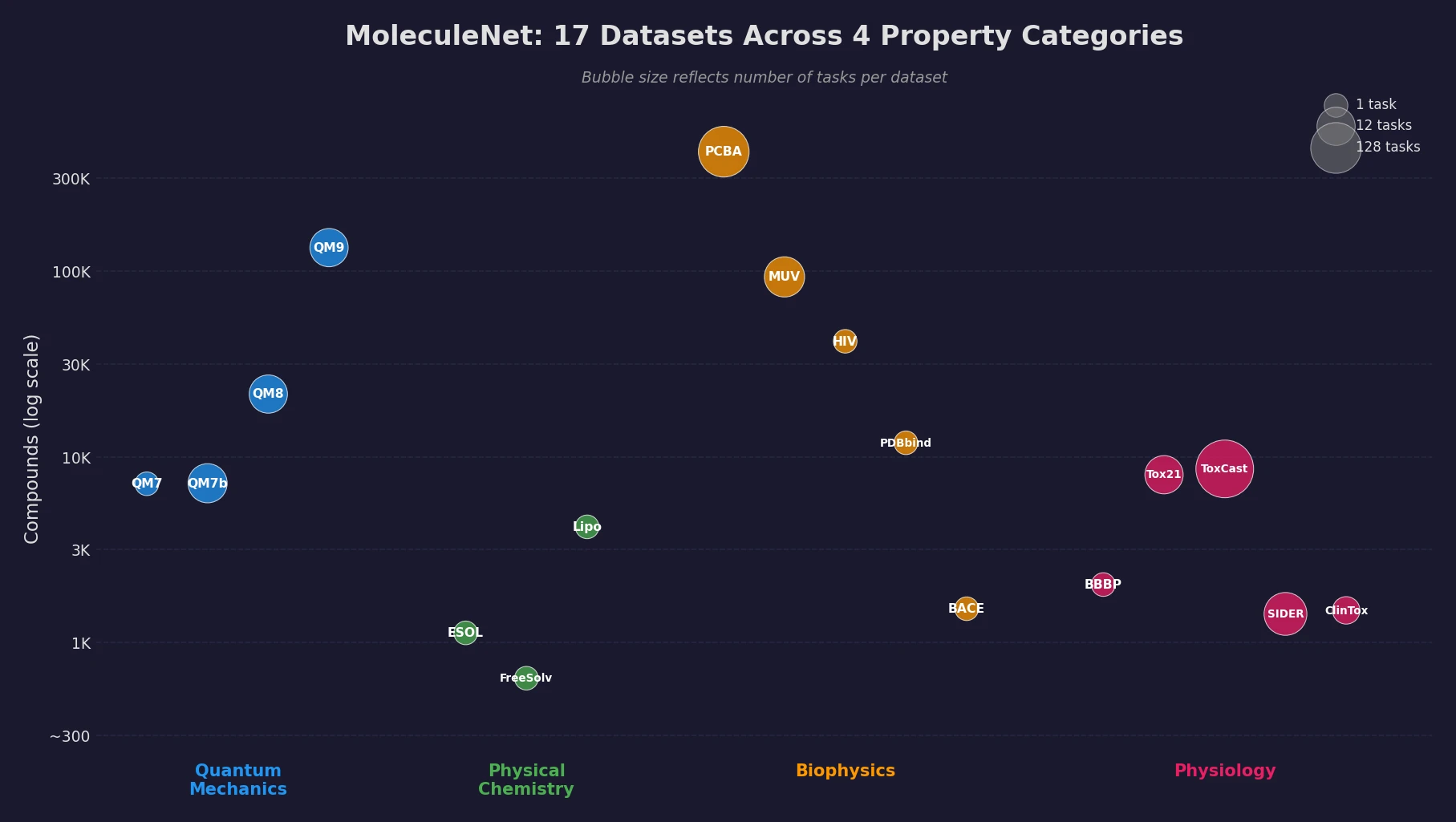
MoleculeNet introduces a large-scale benchmark suite for molecular machine learning, curating over 700,000 compounds across 17 datasets with standardized metrics, data splits, and featurization methods integrated into the DeepChem open-source library.
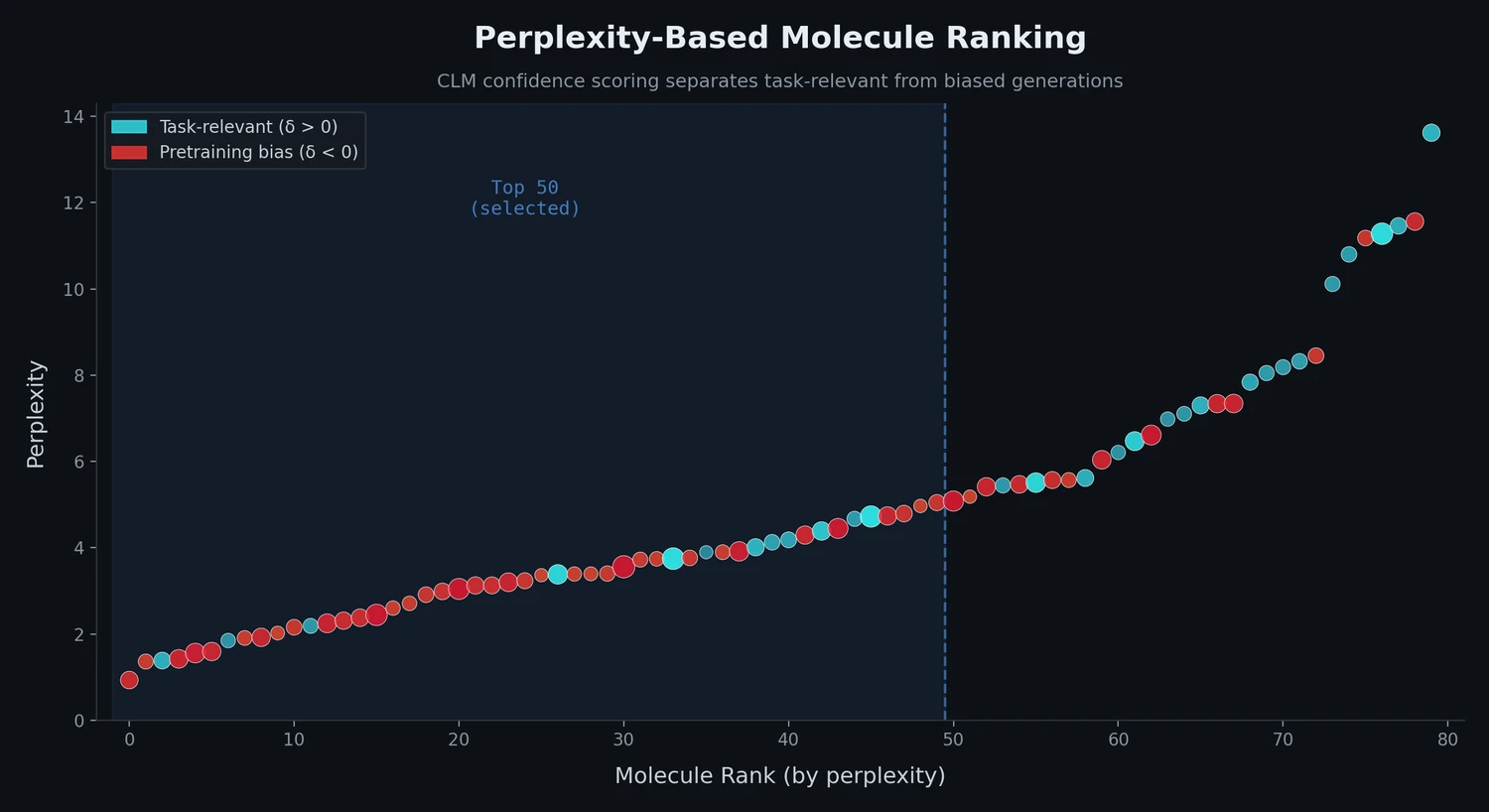
This study applies perplexity, a model-intrinsic metric from NLP, to rank de novo molecular designs generated by SMILES-based chemical language models and introduces a delta score to detect pretraining bias in transfer-learned CLMs.
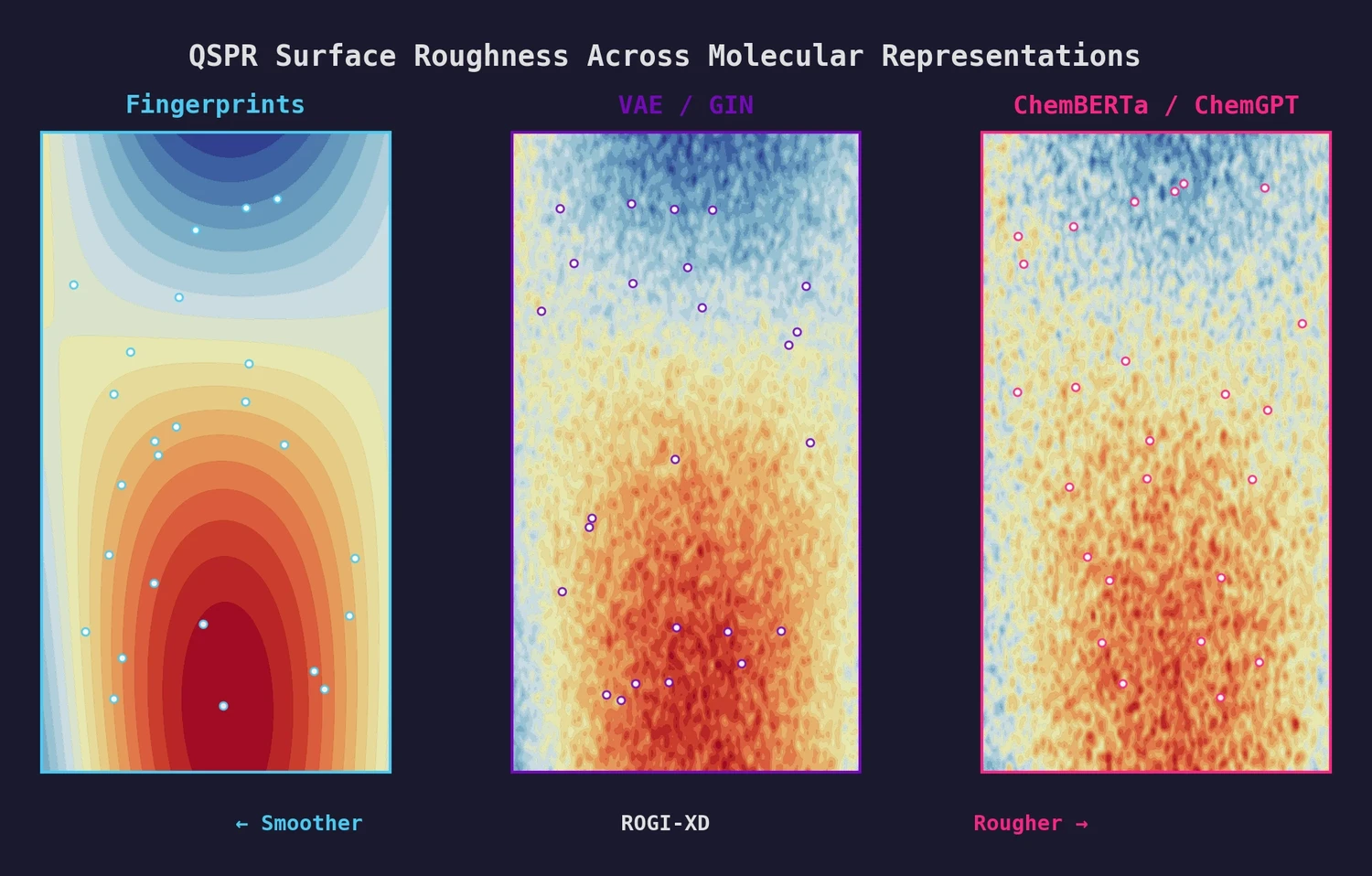
This paper introduces ROGI-XD, a reformulation of the ROuGhness Index that enables fair comparison of QSPR surface roughness across molecular representations of different dimensionalities. Evaluating VAE, GIN, ChemBERTa, and ChemGPT representations, the authors show that pretrained chemical models do not produce smoother structure-property landscapes than simple molecular fingerprints or descriptors.
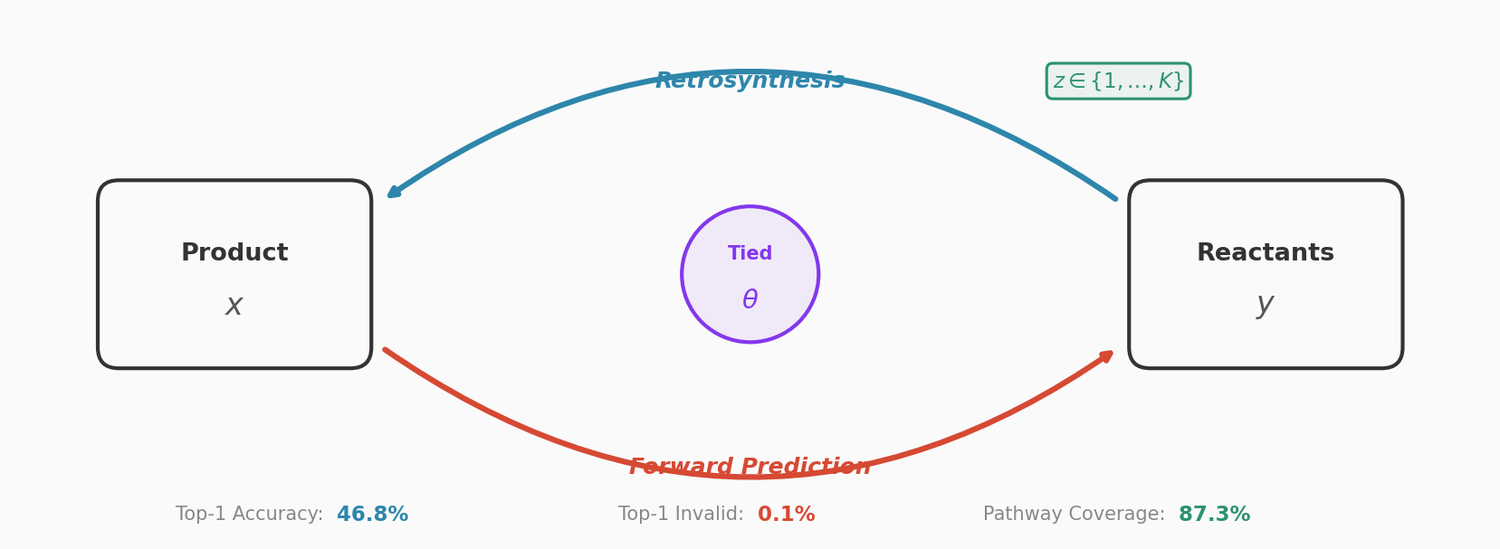
This paper couples a retrosynthesis transformer with a forward reaction transformer through parameter sharing, cycle consistency checks, and multinomial latent variables. The combined approach reduces top-1 SMILES invalidity to 0.1% on USPTO-50K, improves top-10 accuracy to 78.5%, and achieves 87.3% pathway coverage on a multi-pathway in-house dataset.
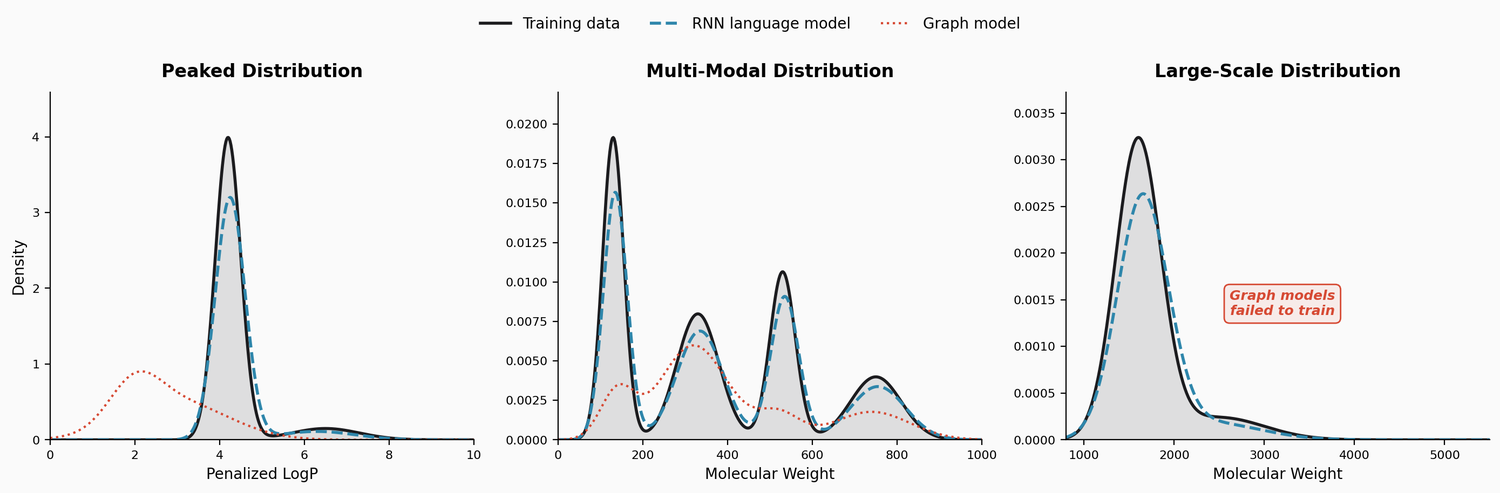
This study benchmarks RNN-based chemical language models against graph generative models on three challenging tasks: high penalized LogP distributions, multi-modal molecular distributions, and large-molecule generation from PubChem. The LSTM language models consistently outperform JTVAE and CGVAE.
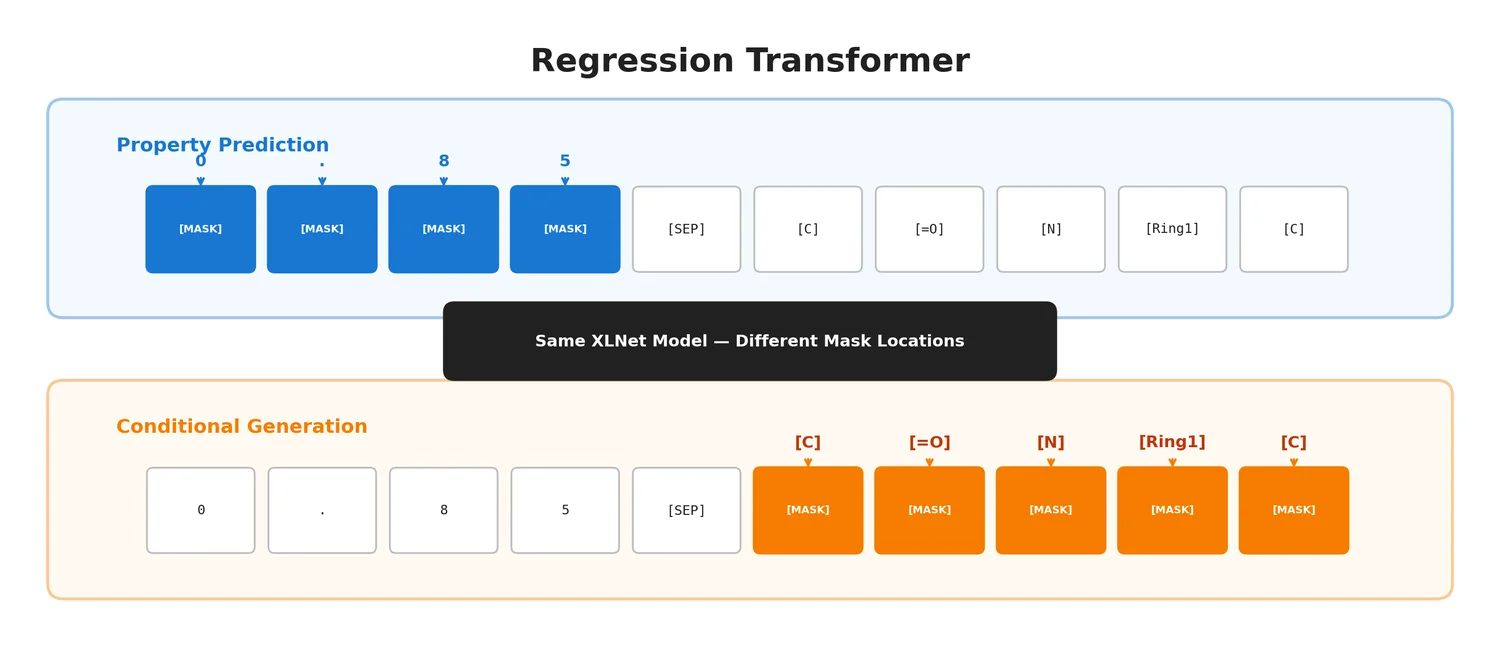
The Regression Transformer (RT) reformulates regression as conditional sequence modelling, enabling a single XLNet-based model to both predict continuous molecular properties and generate novel molecules conditioned on desired property values.
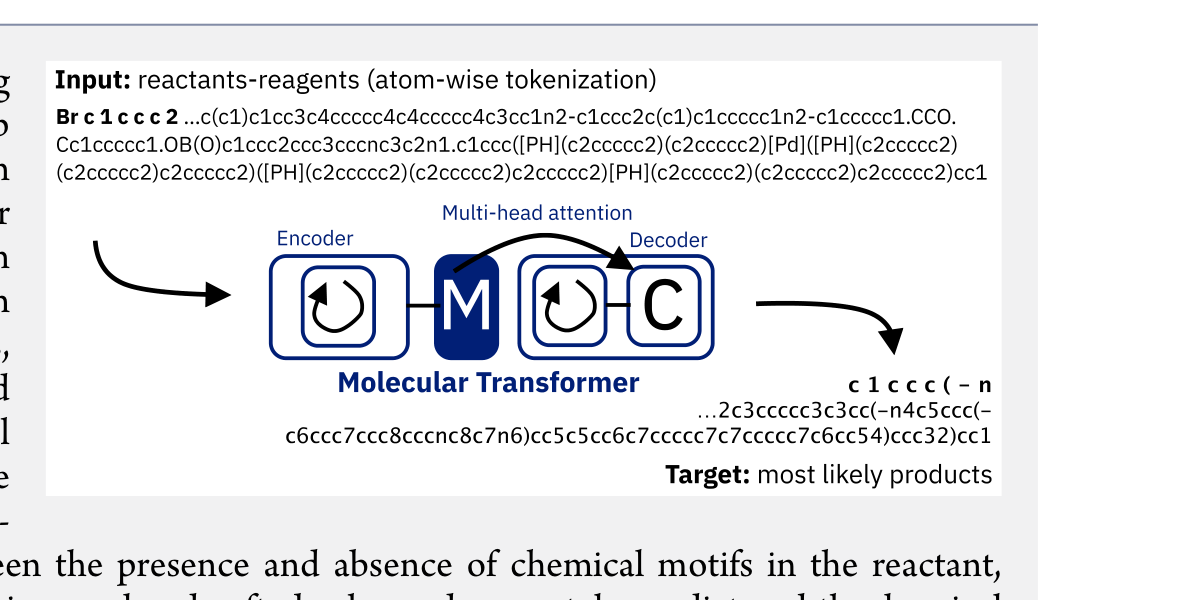
The Molecular Transformer applies the Transformer architecture to forward reaction prediction, treating it as SMILES-to-SMILES machine translation. It achieves 90.4% top-1 accuracy on USPTO_MIT, outperforms quantum-chemistry baselines on regioselectivity, and provides calibrated uncertainty scores (0.89 AUC-ROC) for ranking synthesis pathways.