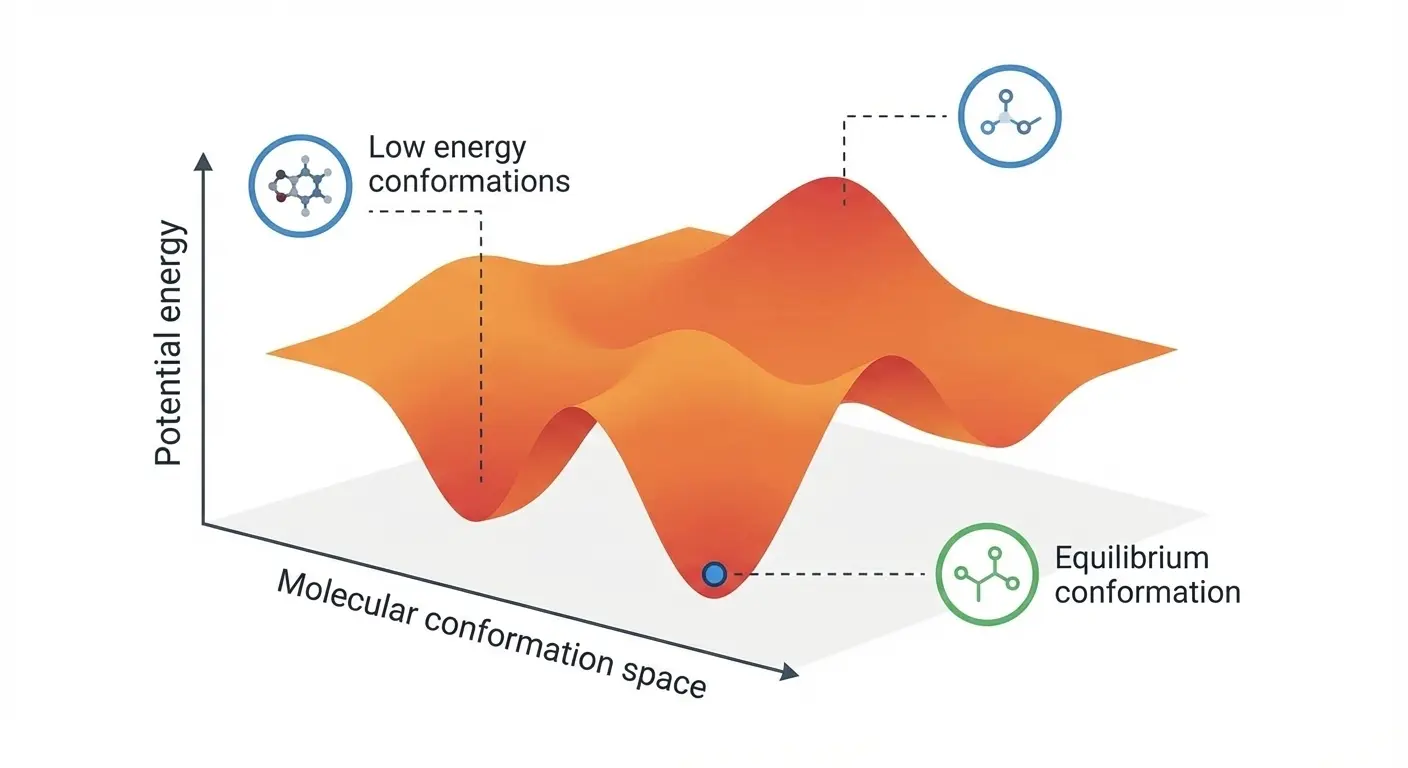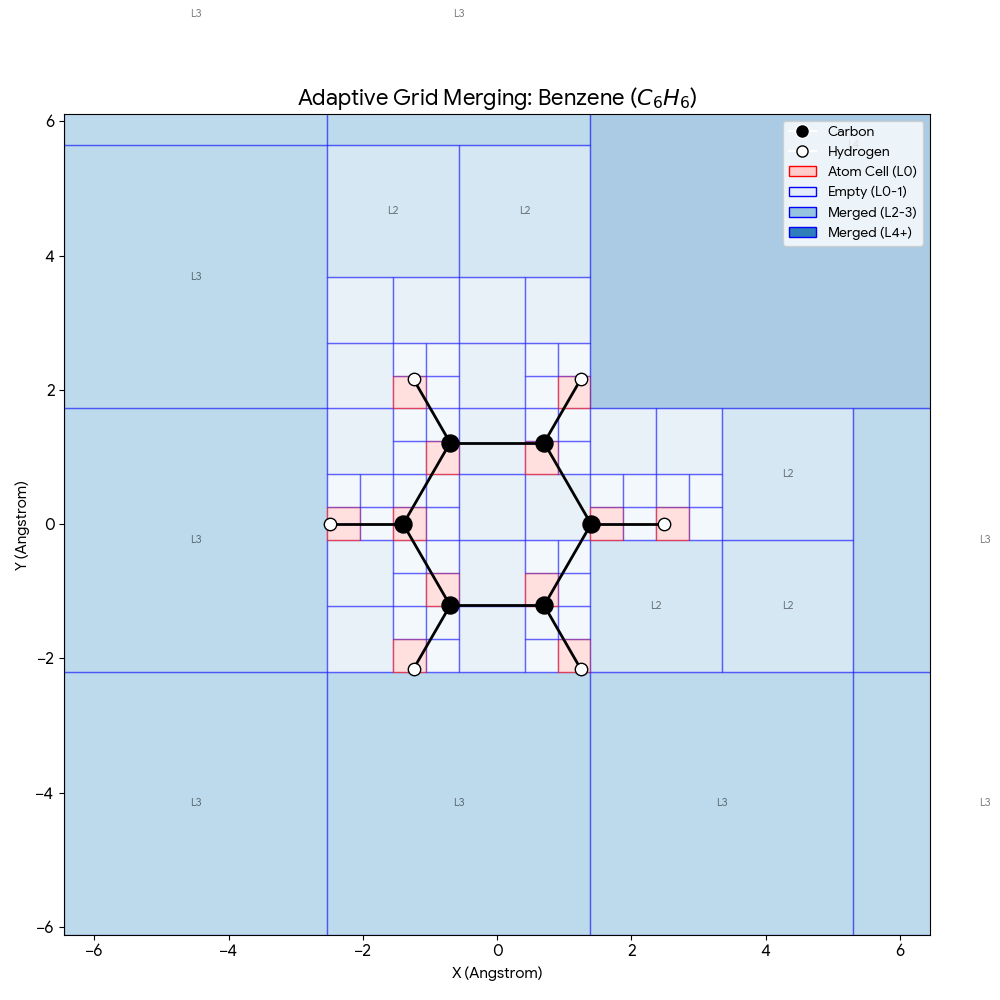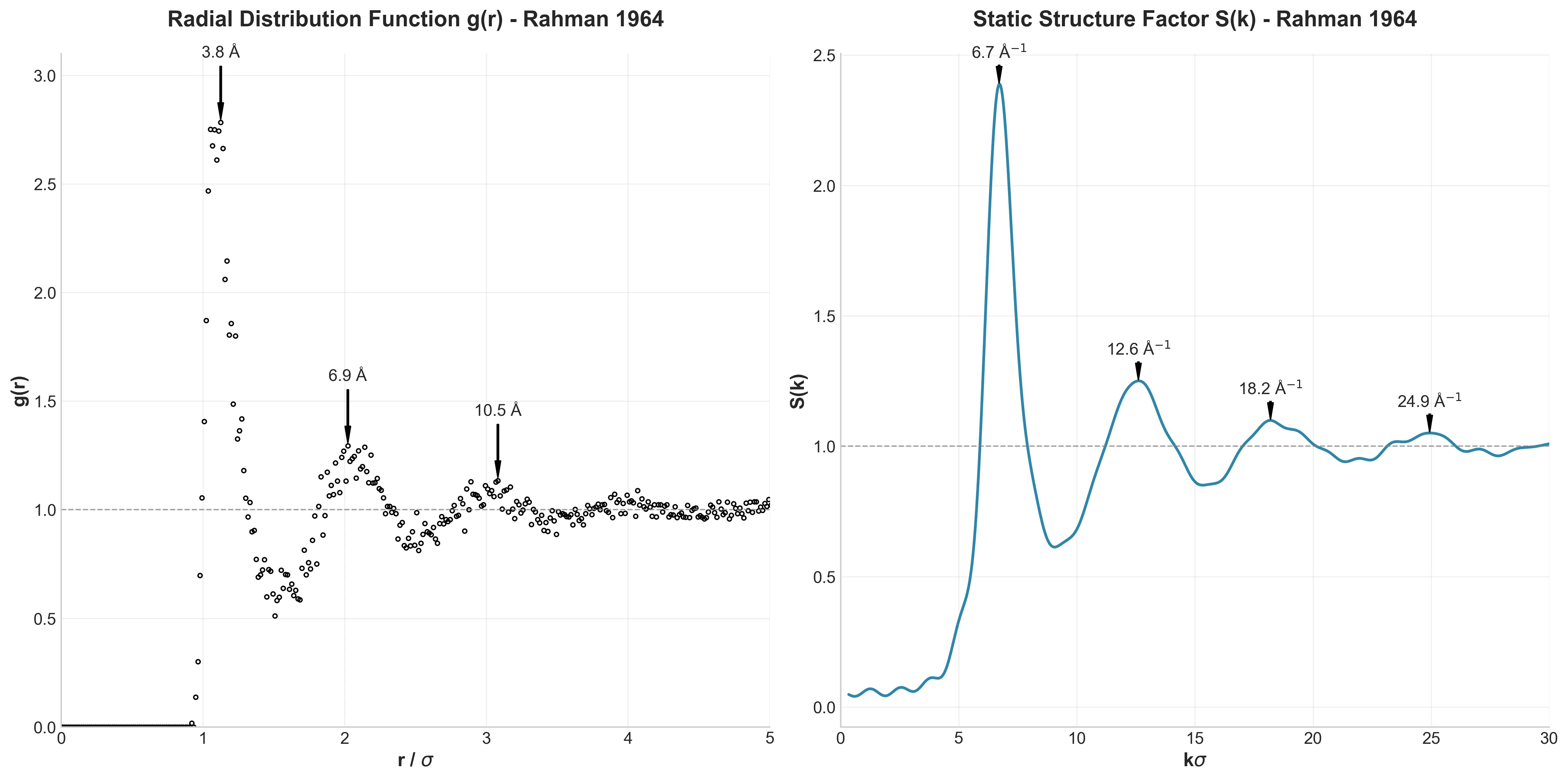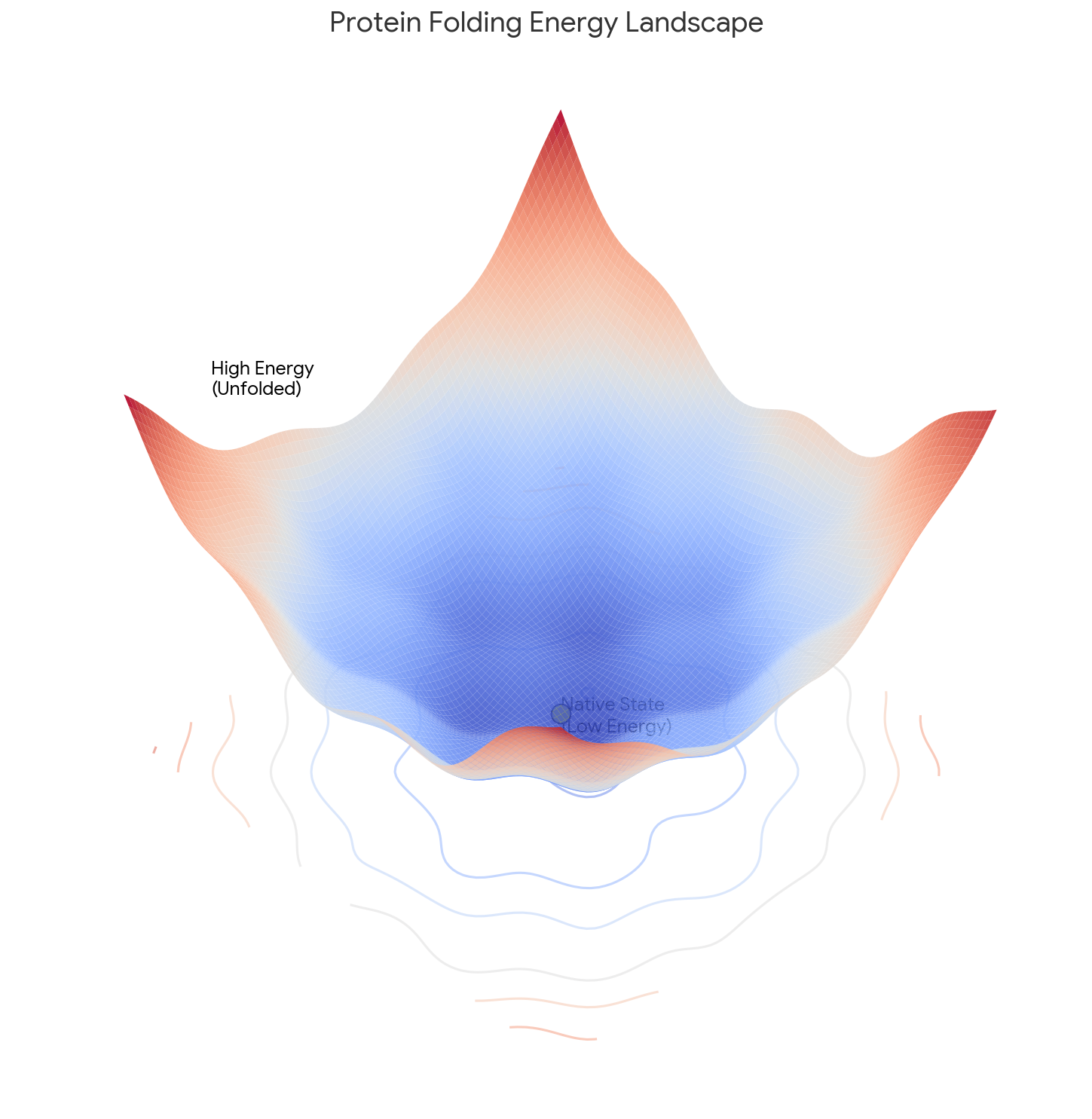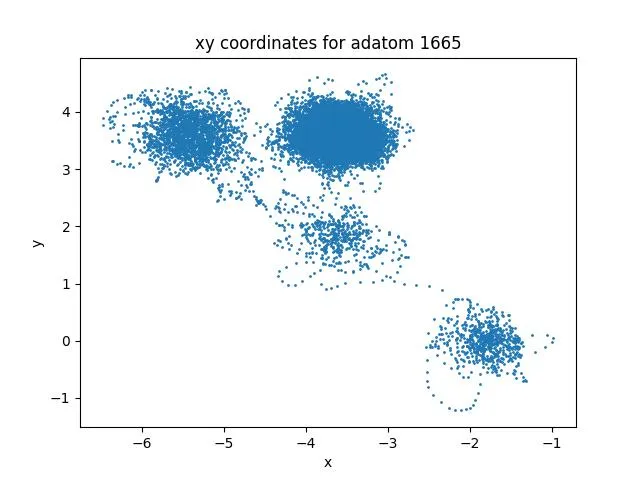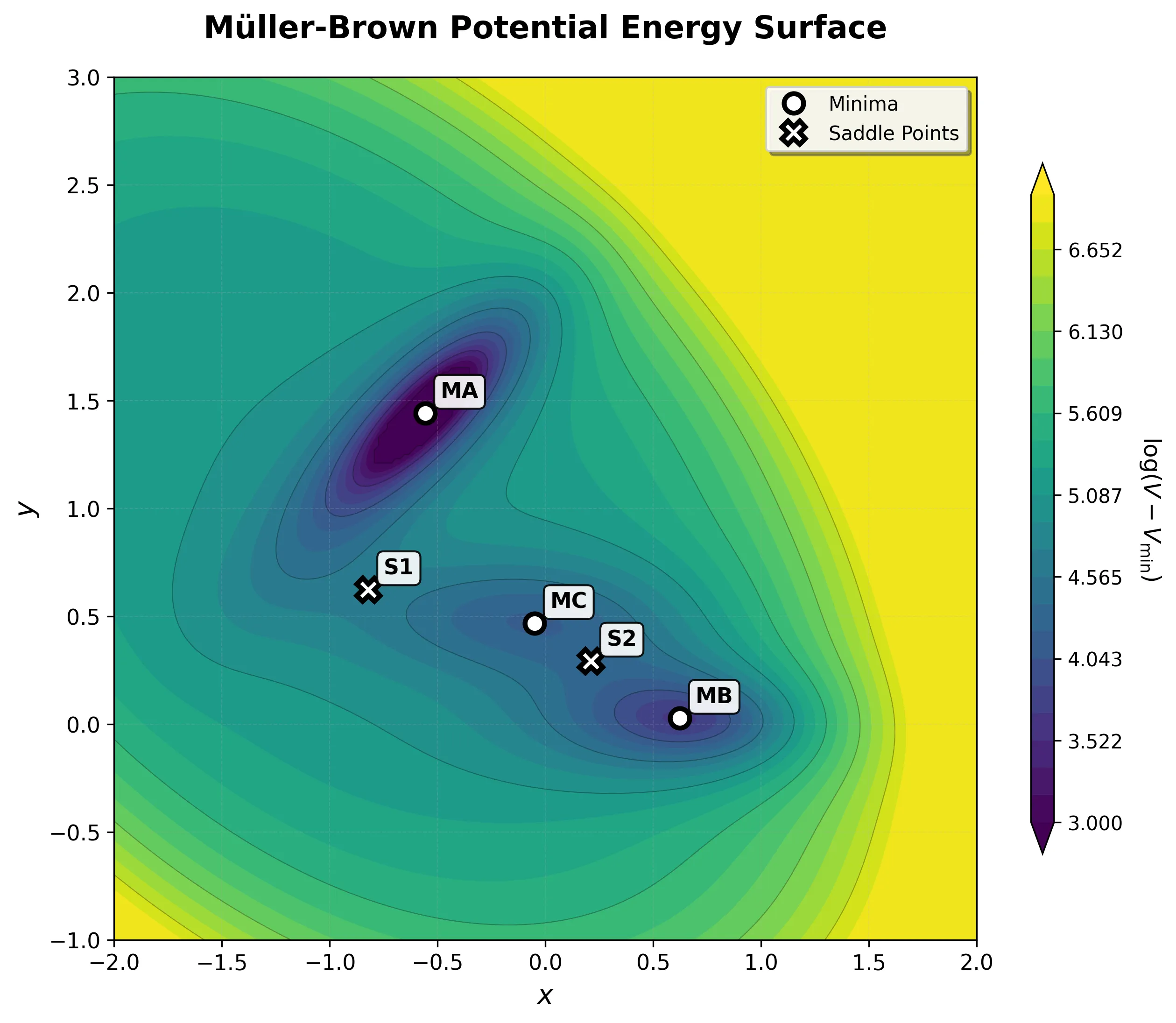
Müller-Brown Transition: Langevin Dynamics Simulation
Experience rare transition events between energy basins in this extended Müller-Brown simulation. Watch as particles overcome energy barriers to explore different regions of the potential energy landscape.