In Situ XRD of Oxidation-Reduction Oscillations on Pt/SiO2
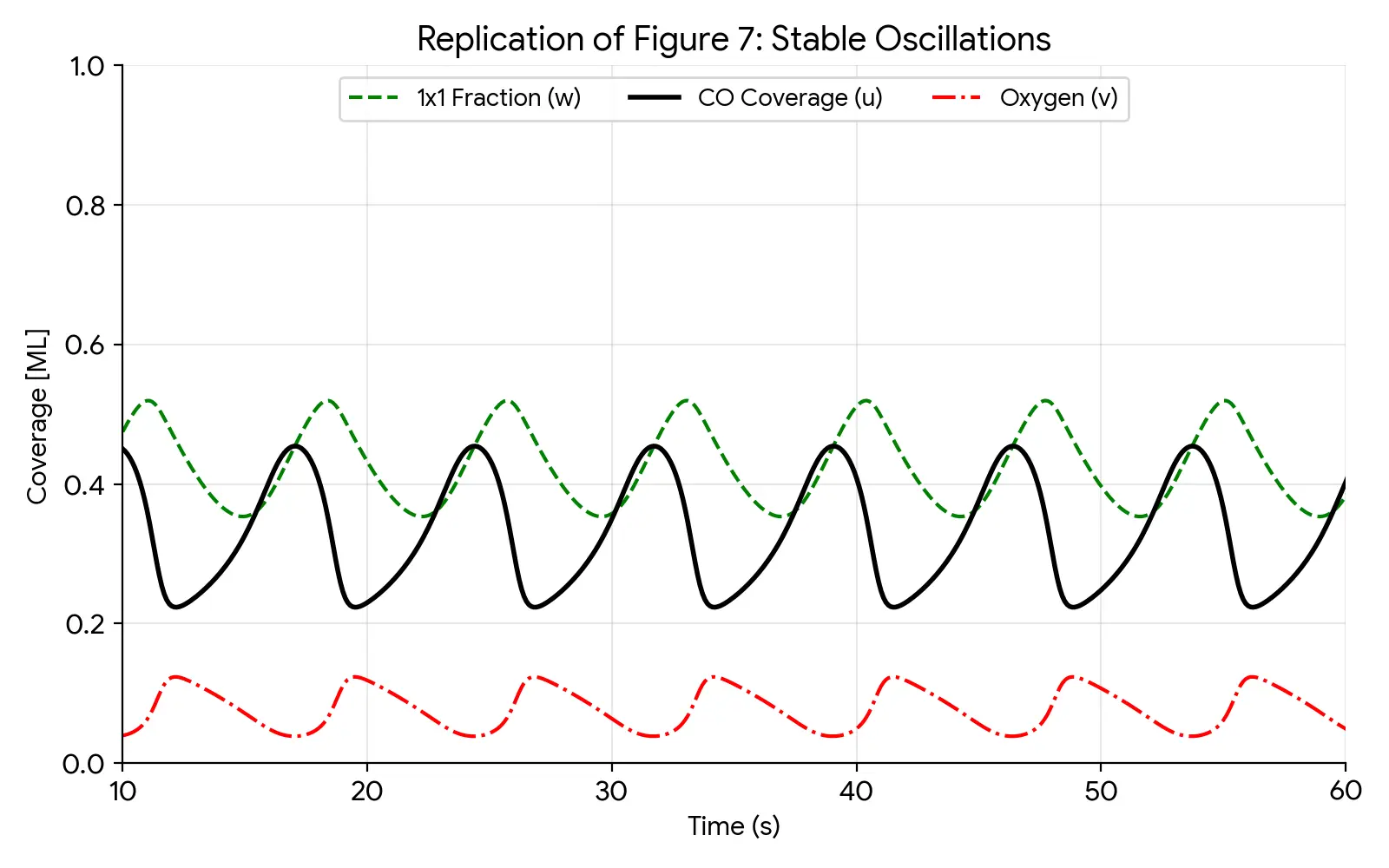
This study provides the first direct experimental proof that rate oscillations in catalytic CO oxidation on supported Pt are driven by a periodic oxidation and reduction of the catalyst surface. By monitoring Bragg peak intensities in situ, the authors confirm the ‘oxide model’ over competing reconstruction or carbon models.