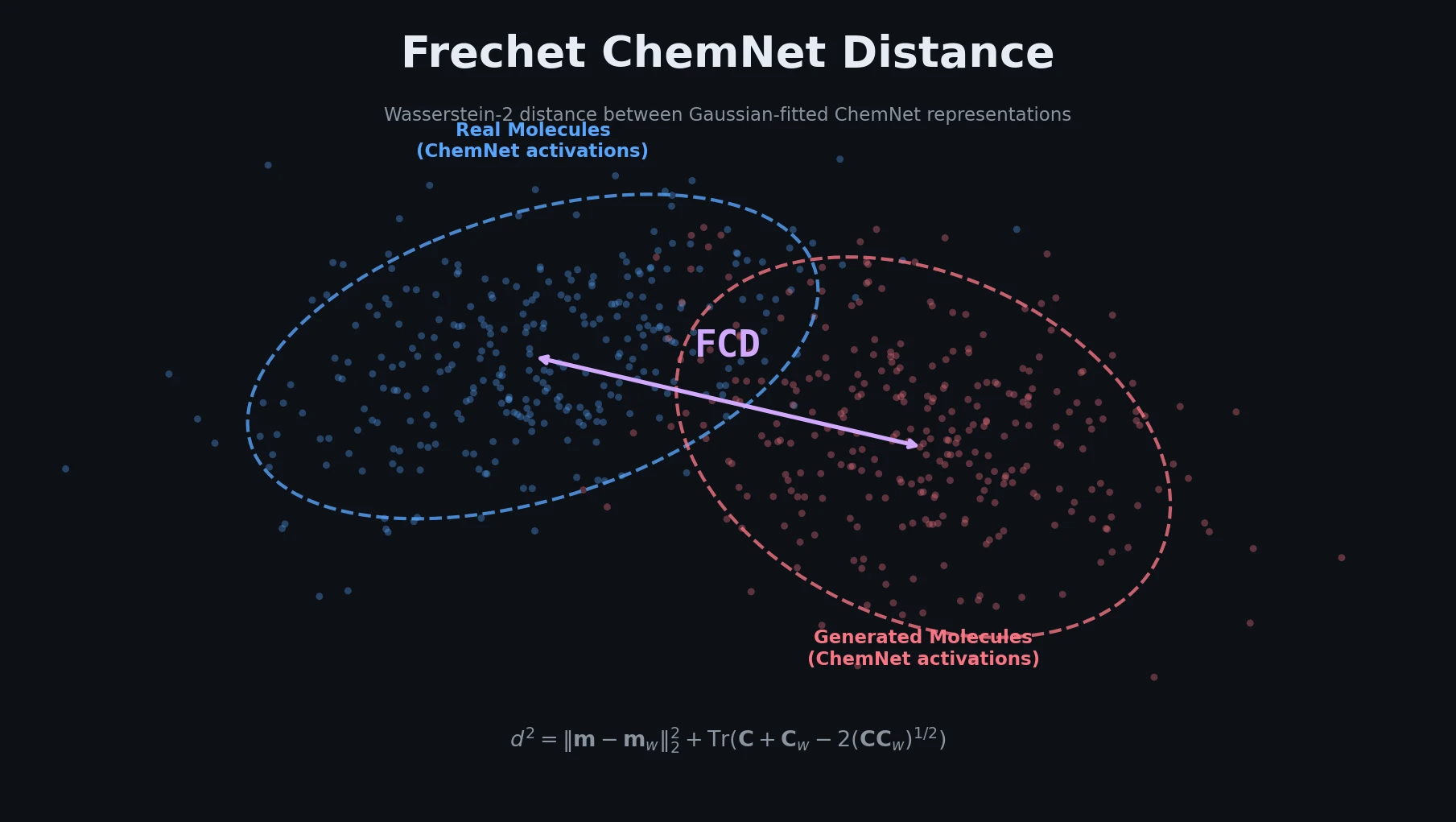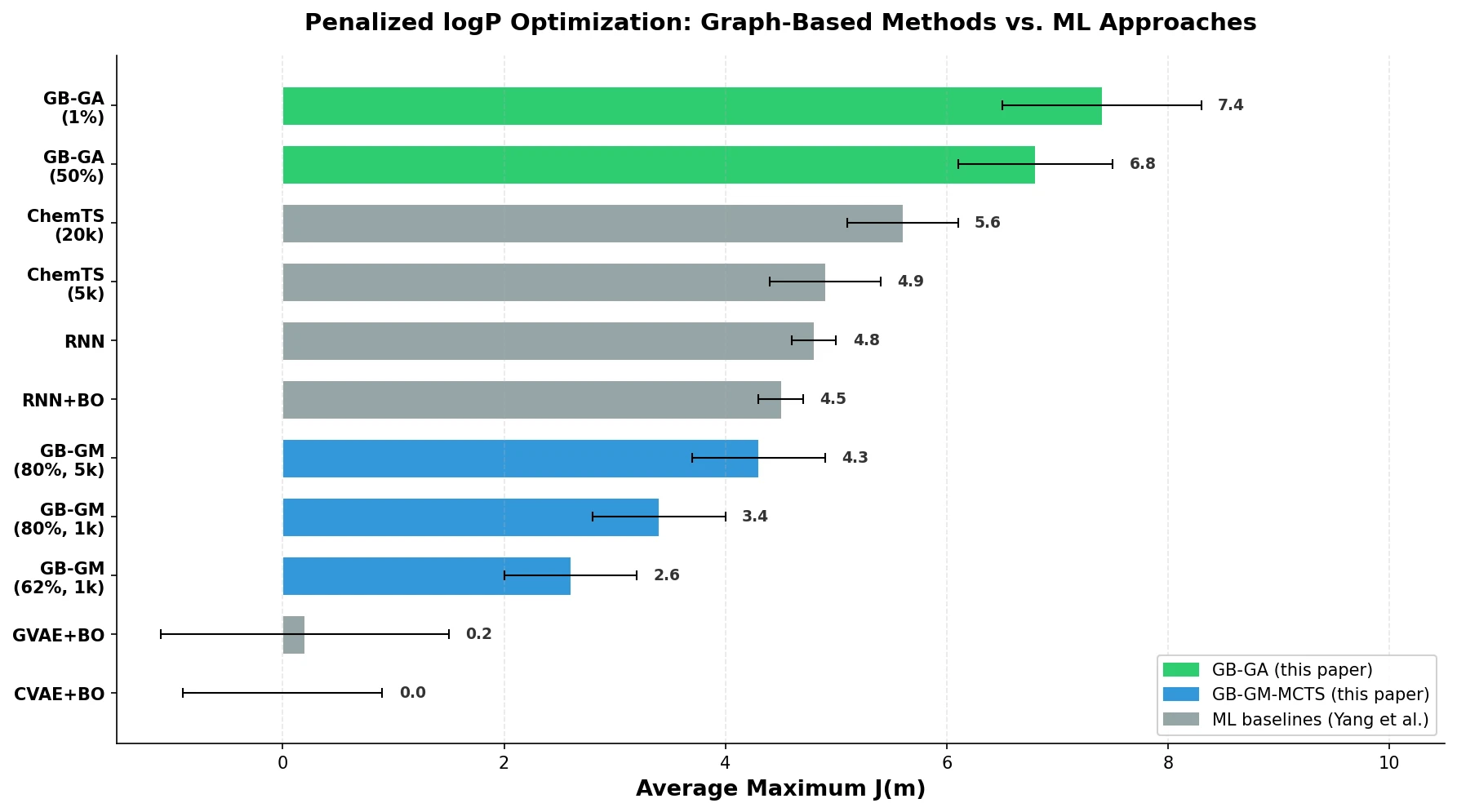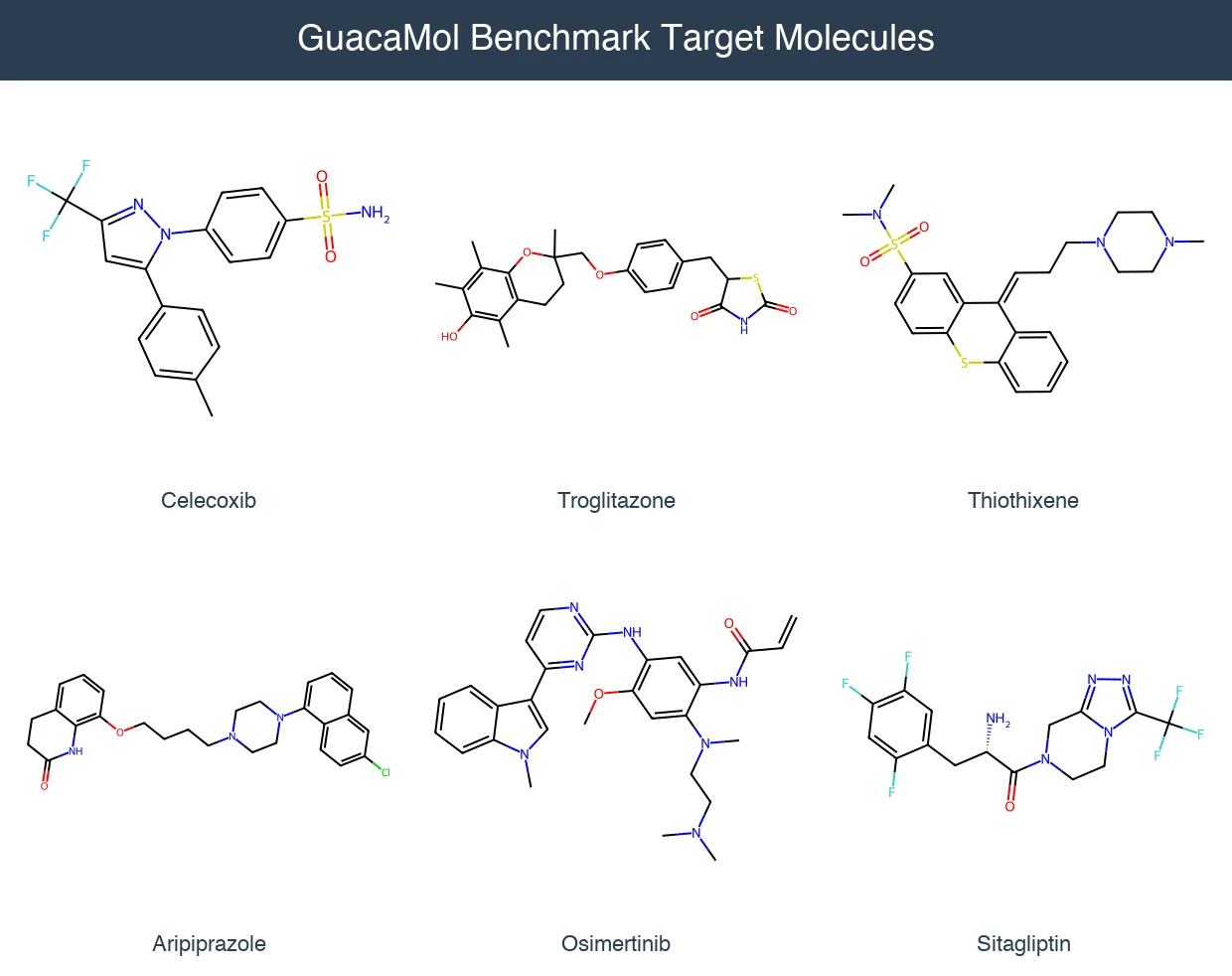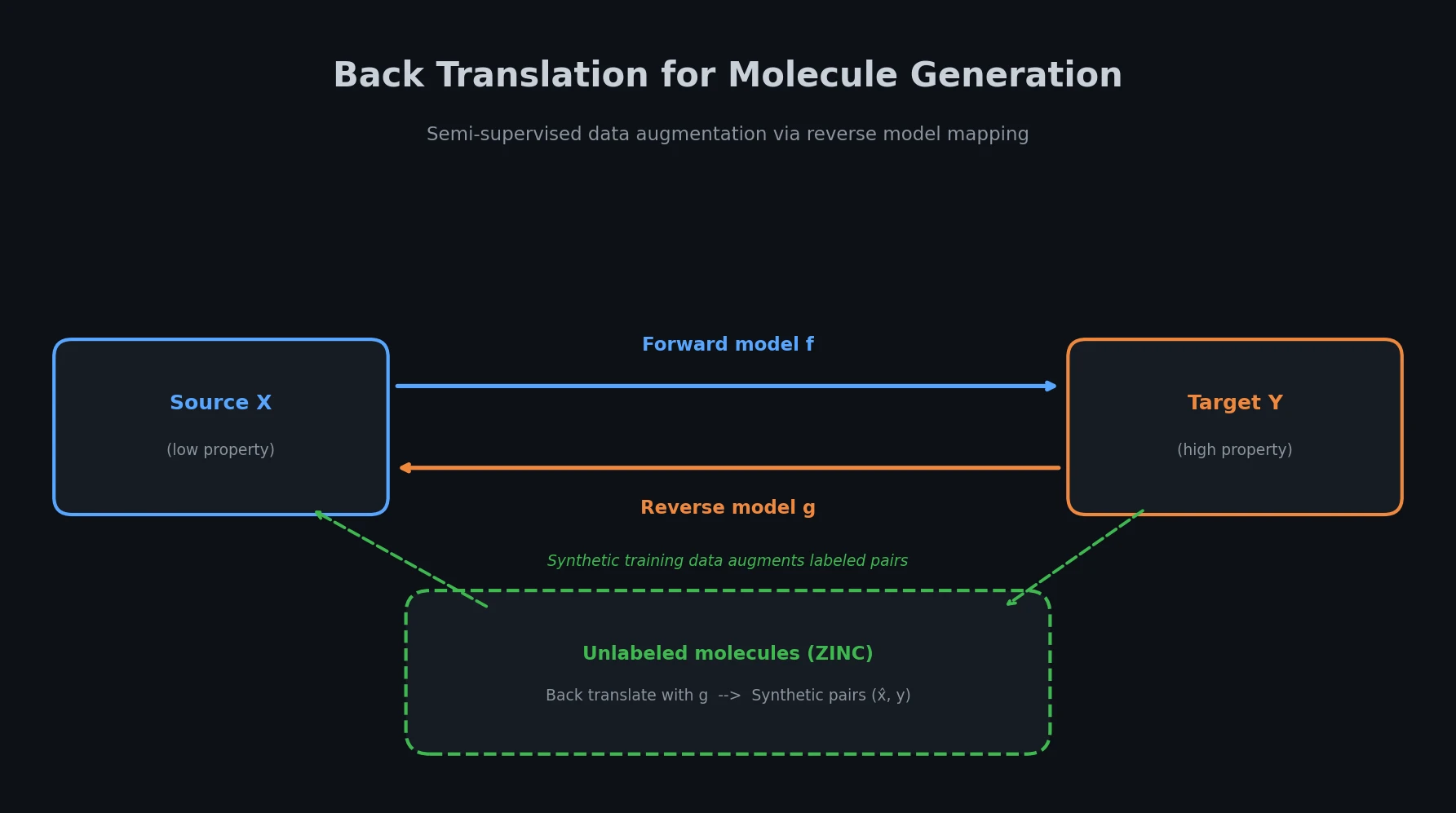
Back Translation for Semi-Supervised Molecule Generation
Adapts back translation from NLP to molecular generation, using unlabeled molecules from ZINC to create synthetic training pairs that improve property optimization and retrosynthesis prediction across Transformer and graph-based architectures.