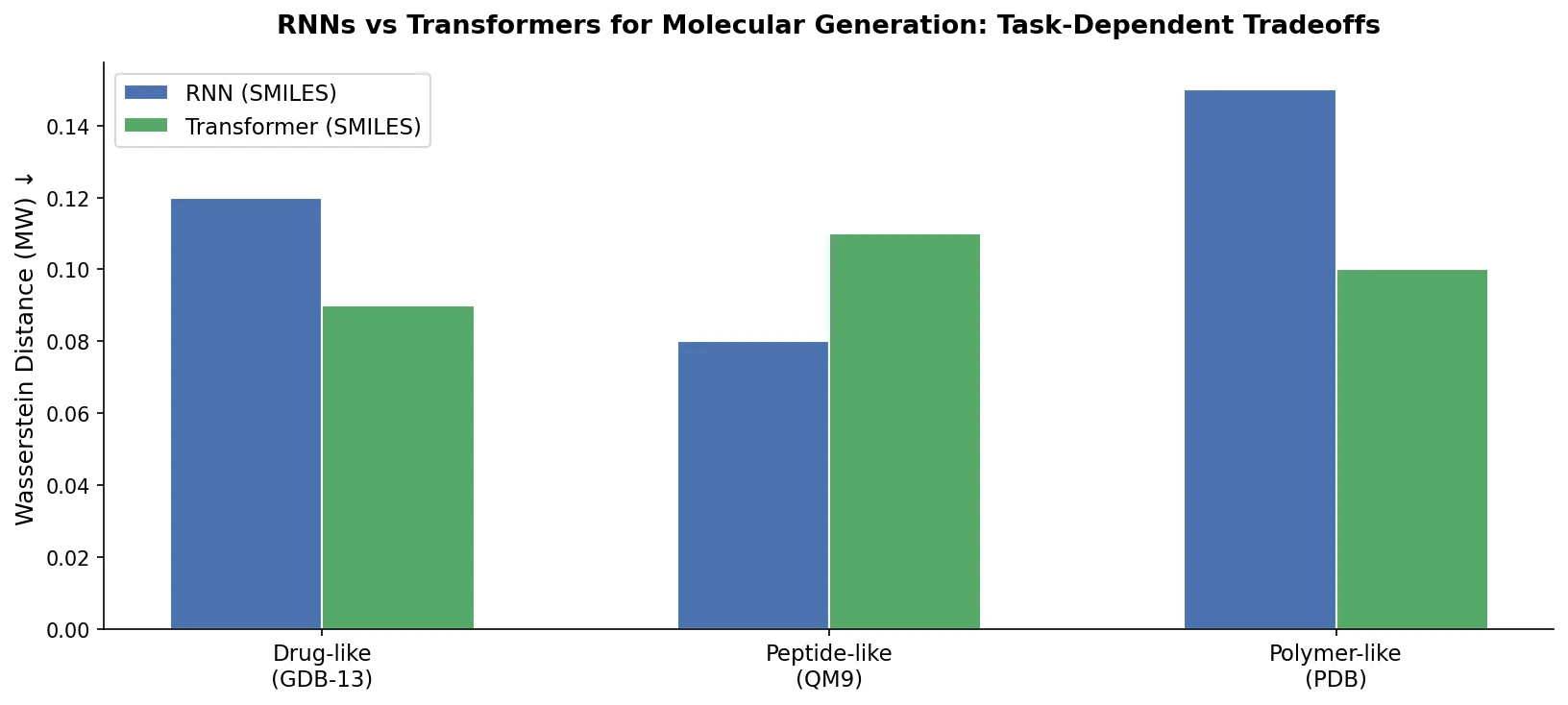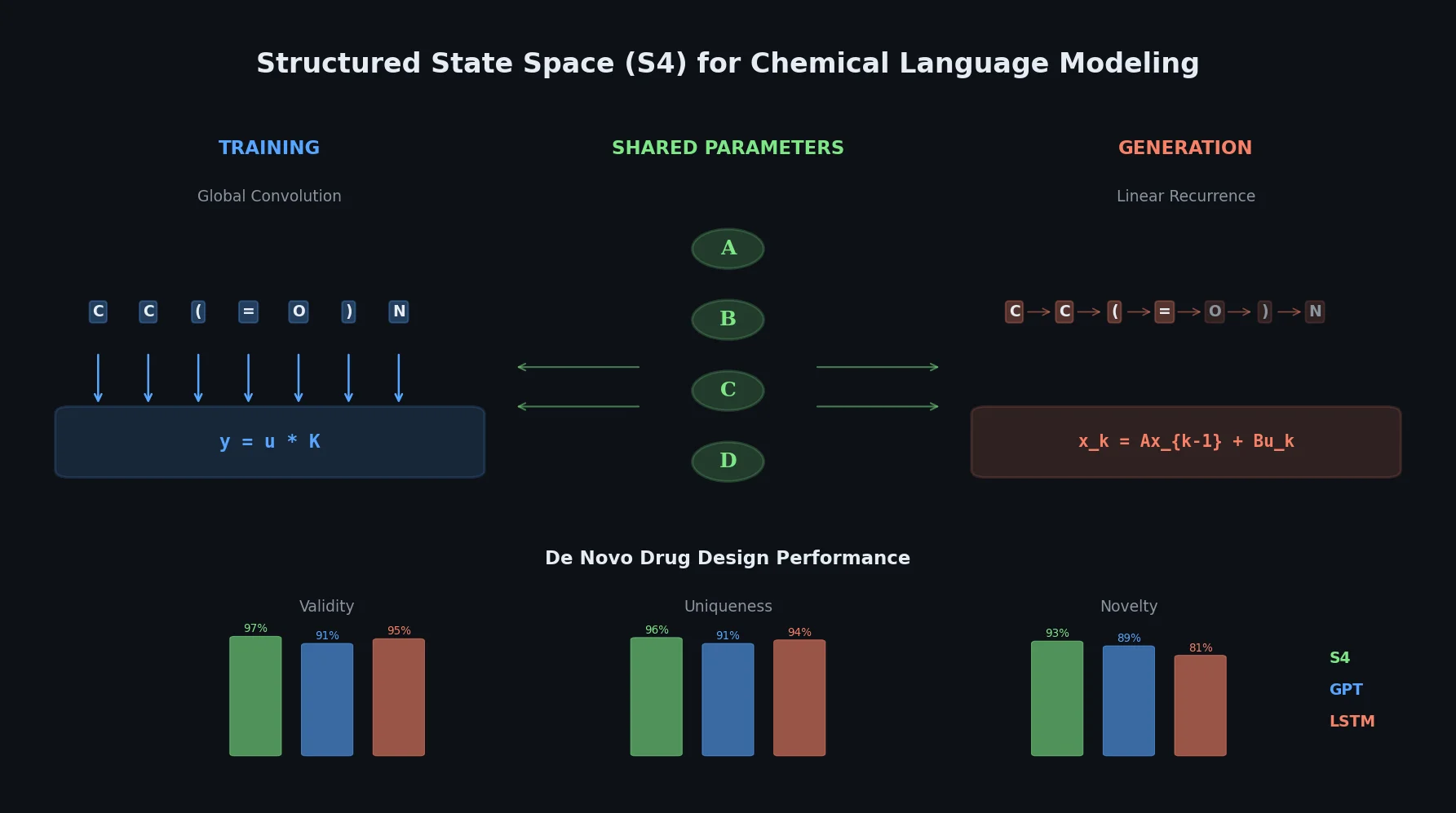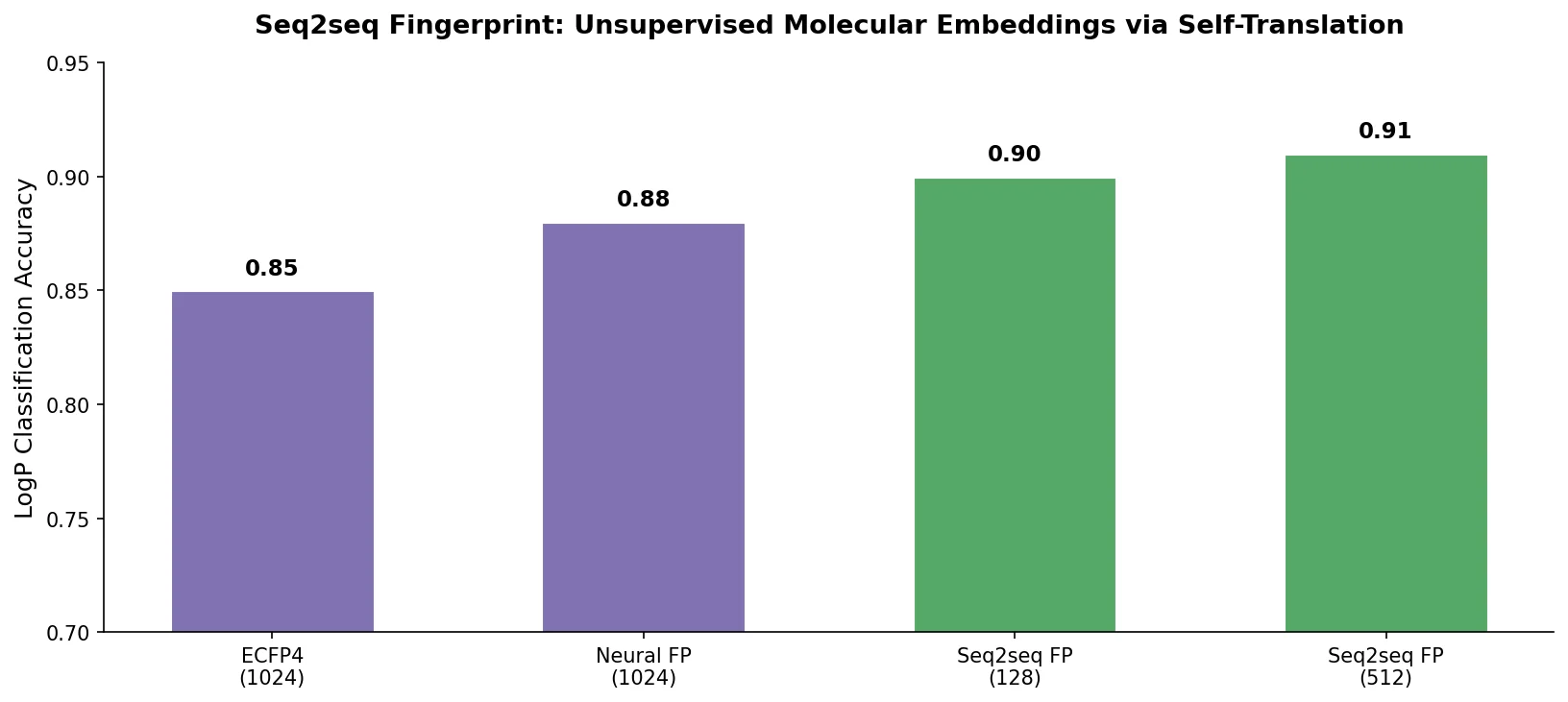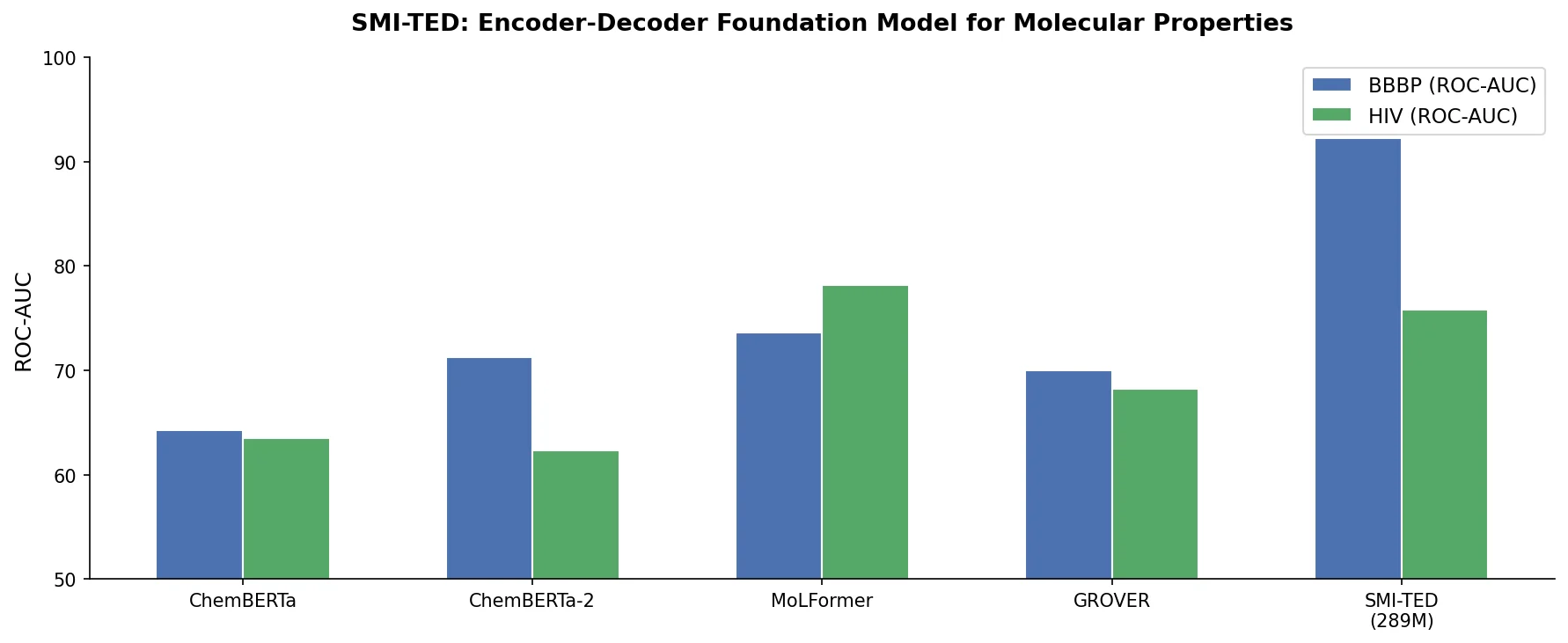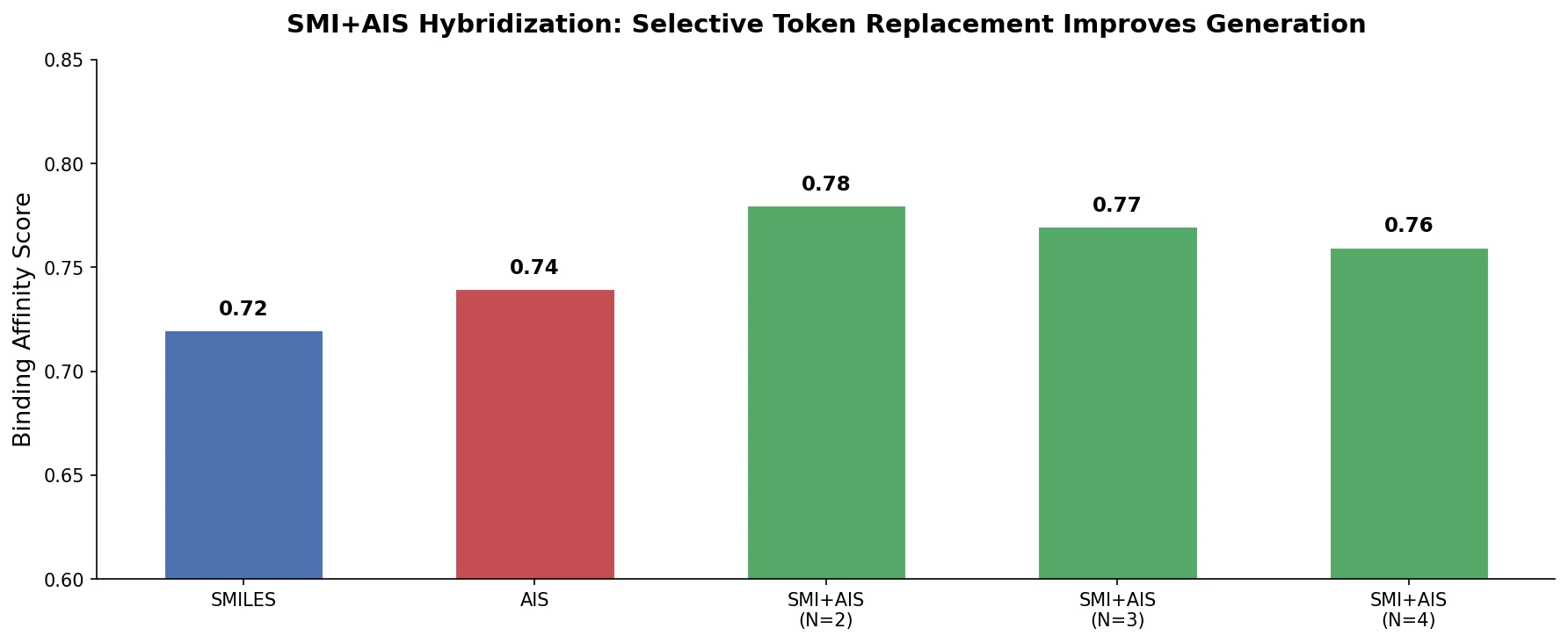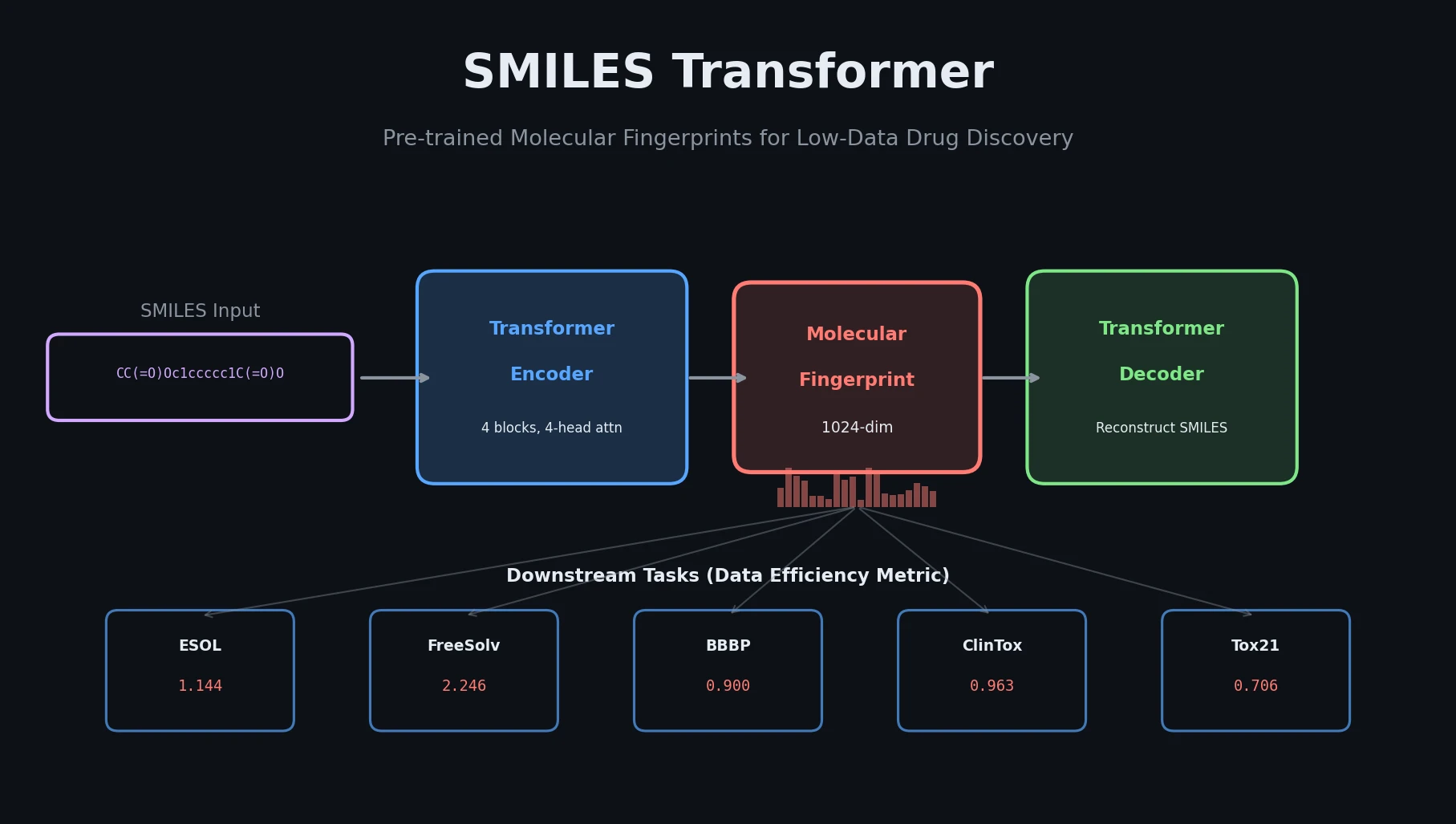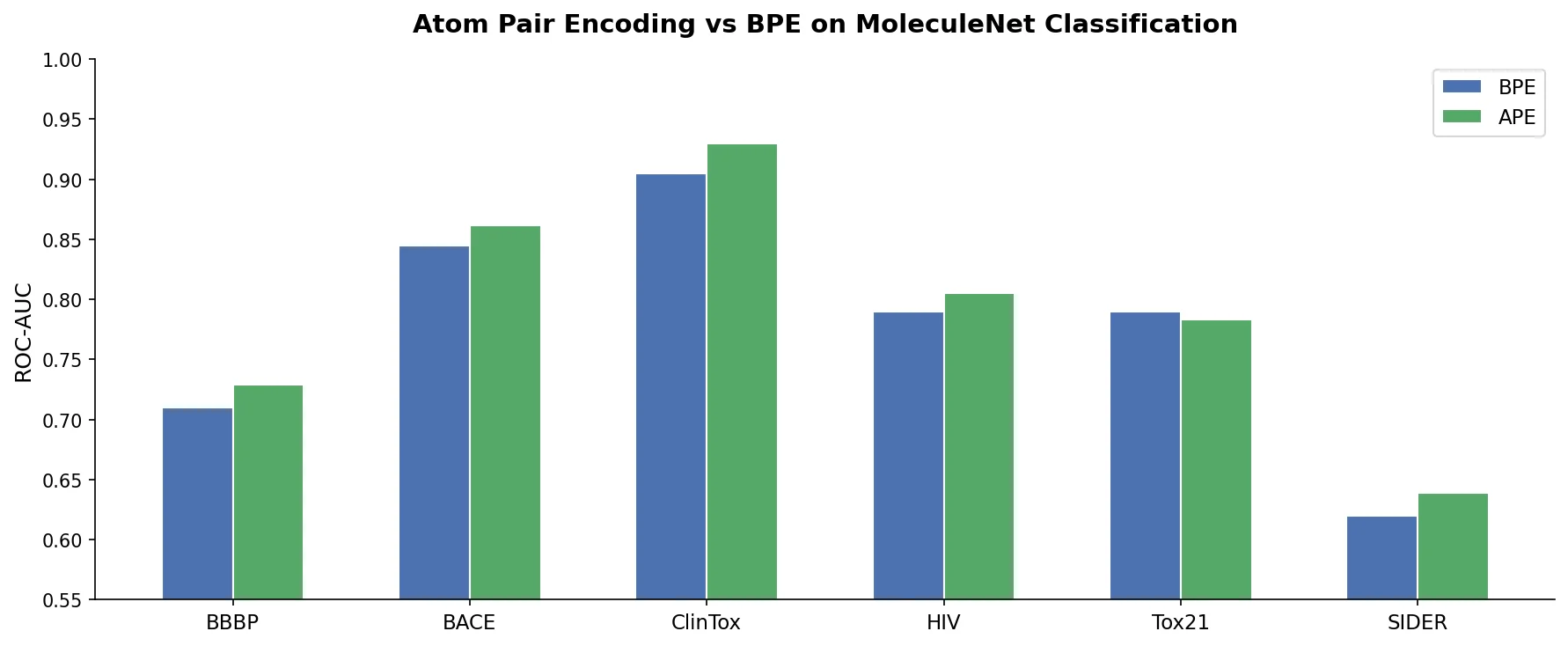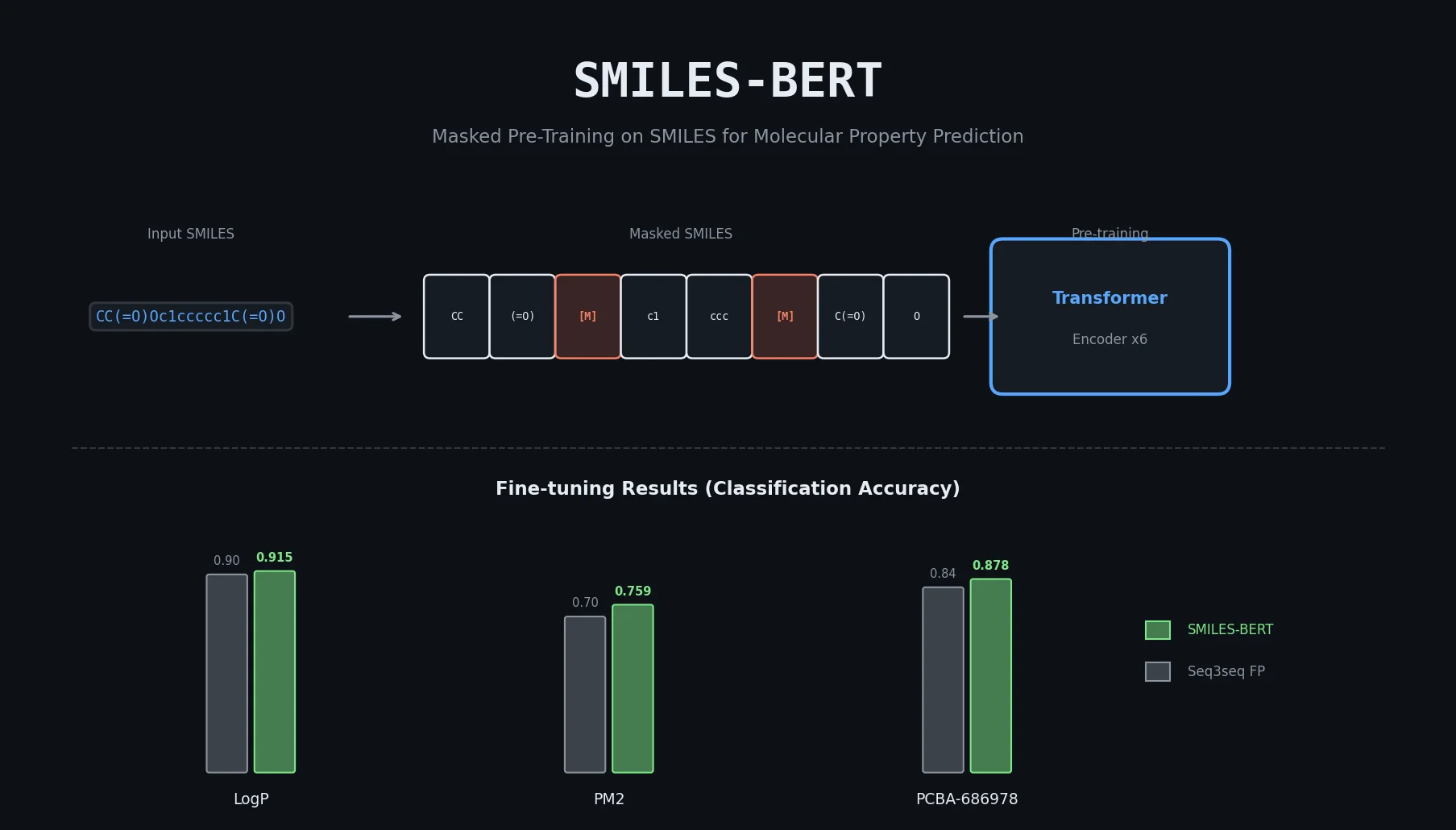
Review: Deep Learning for Molecular Design (2019)
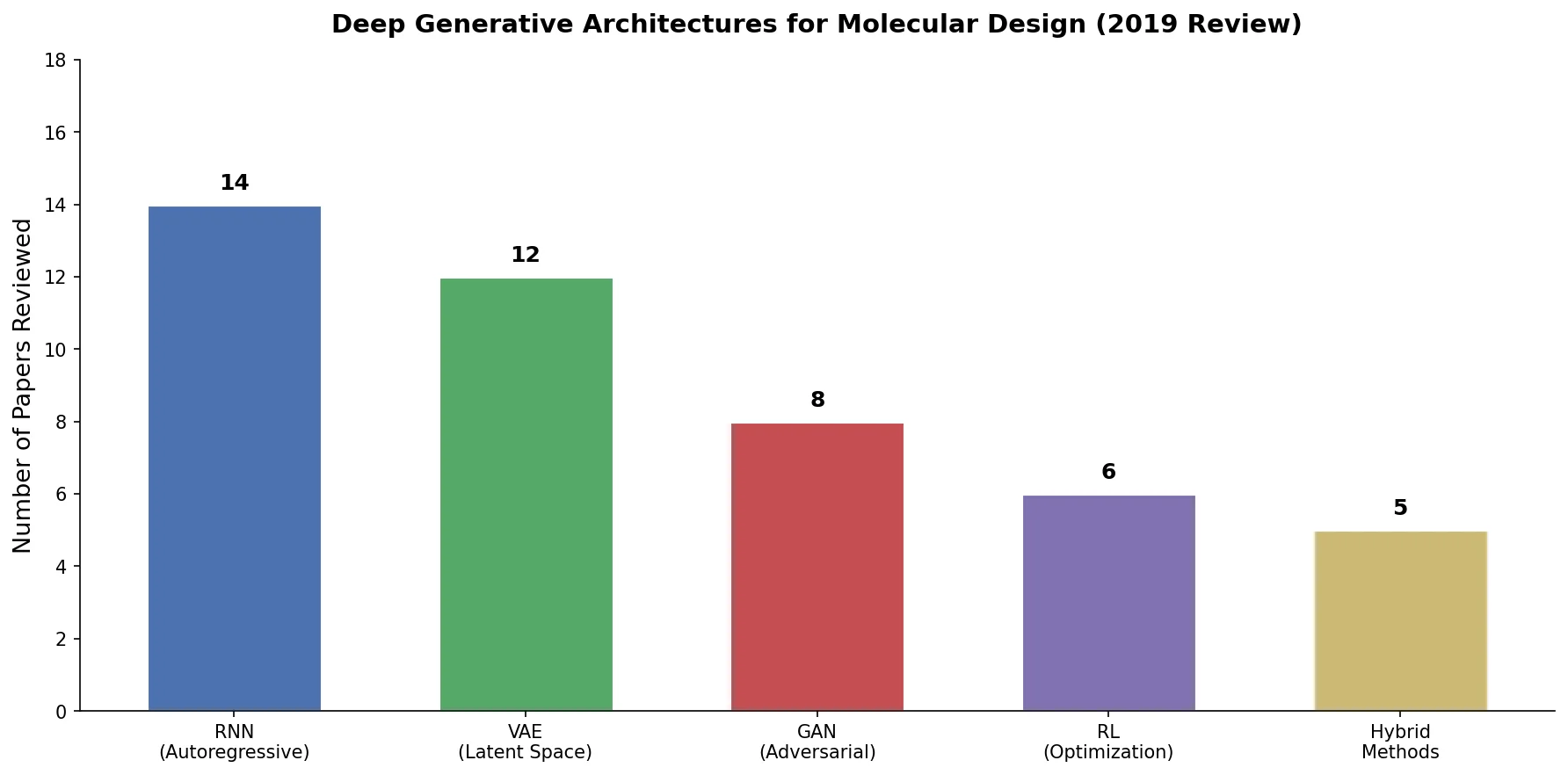
An early and influential review cataloging 45 papers on deep generative modeling for molecules, comparing RNN, VAE, GAN, and reinforcement learning architectures across SMILES and graph-based representations.