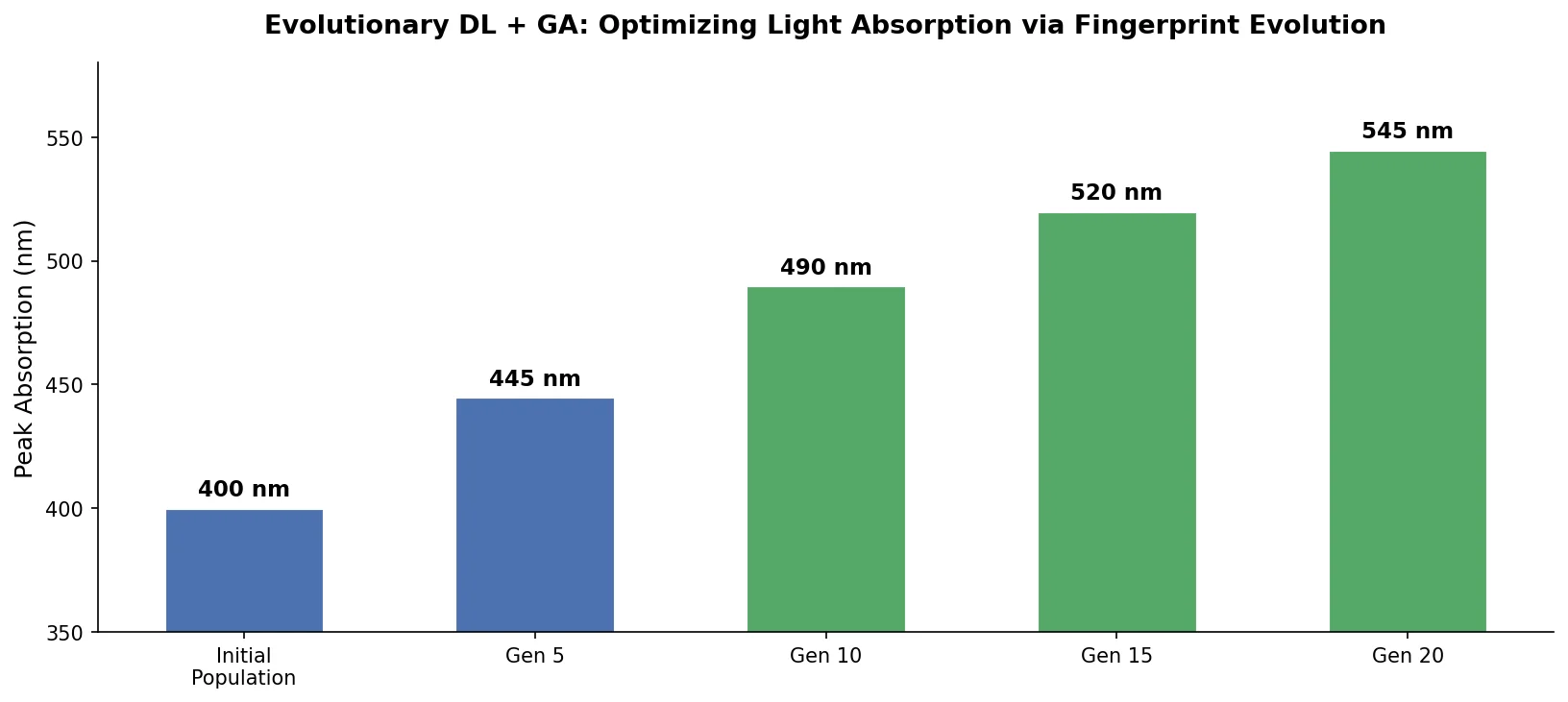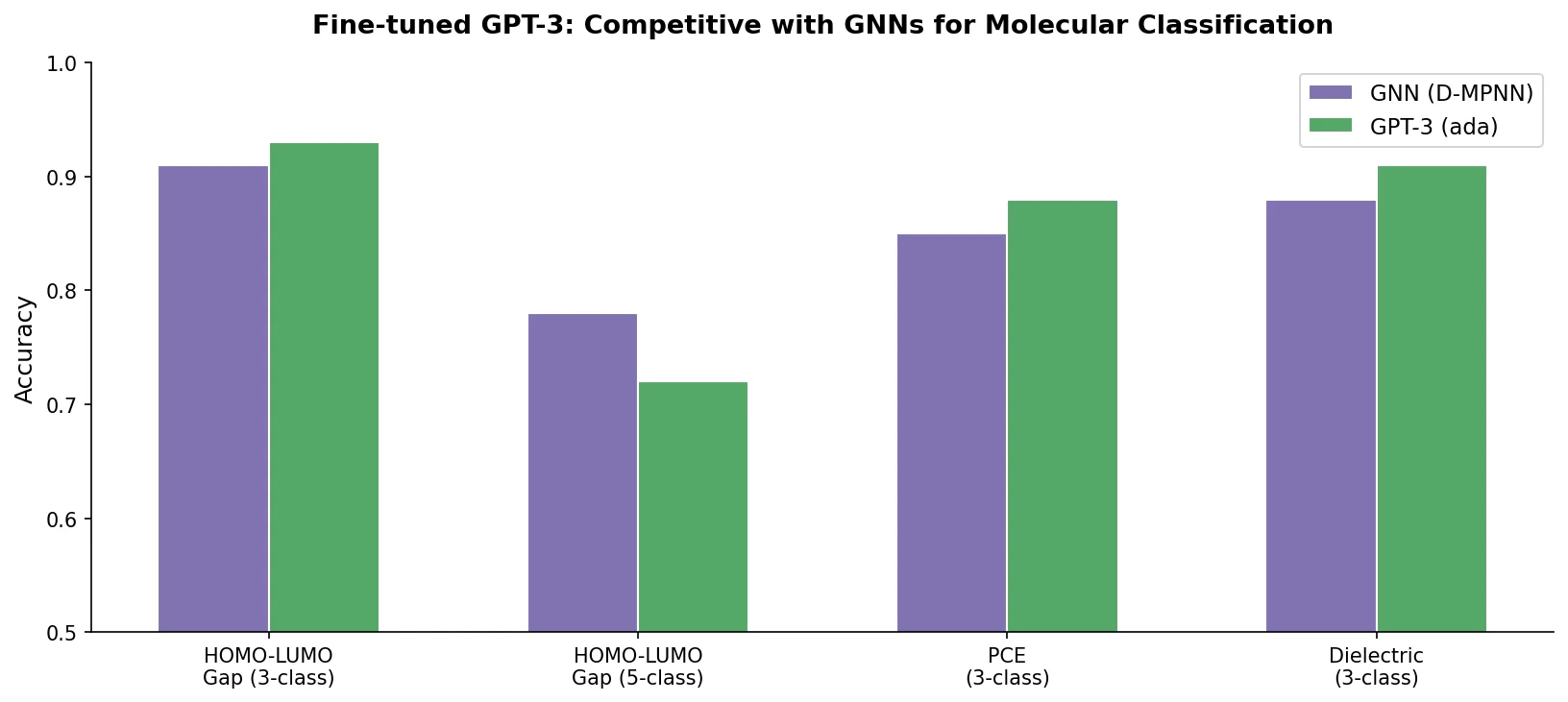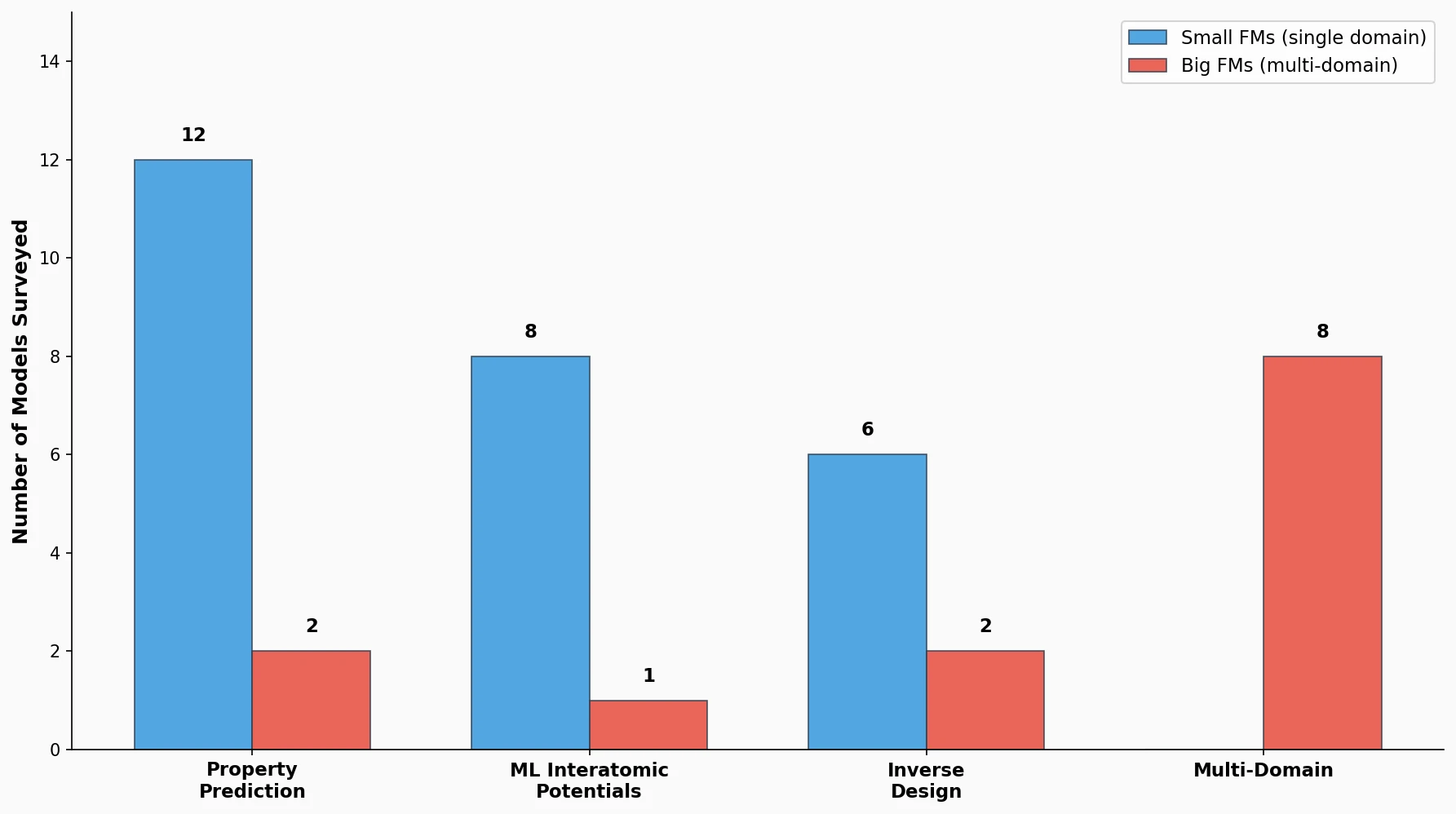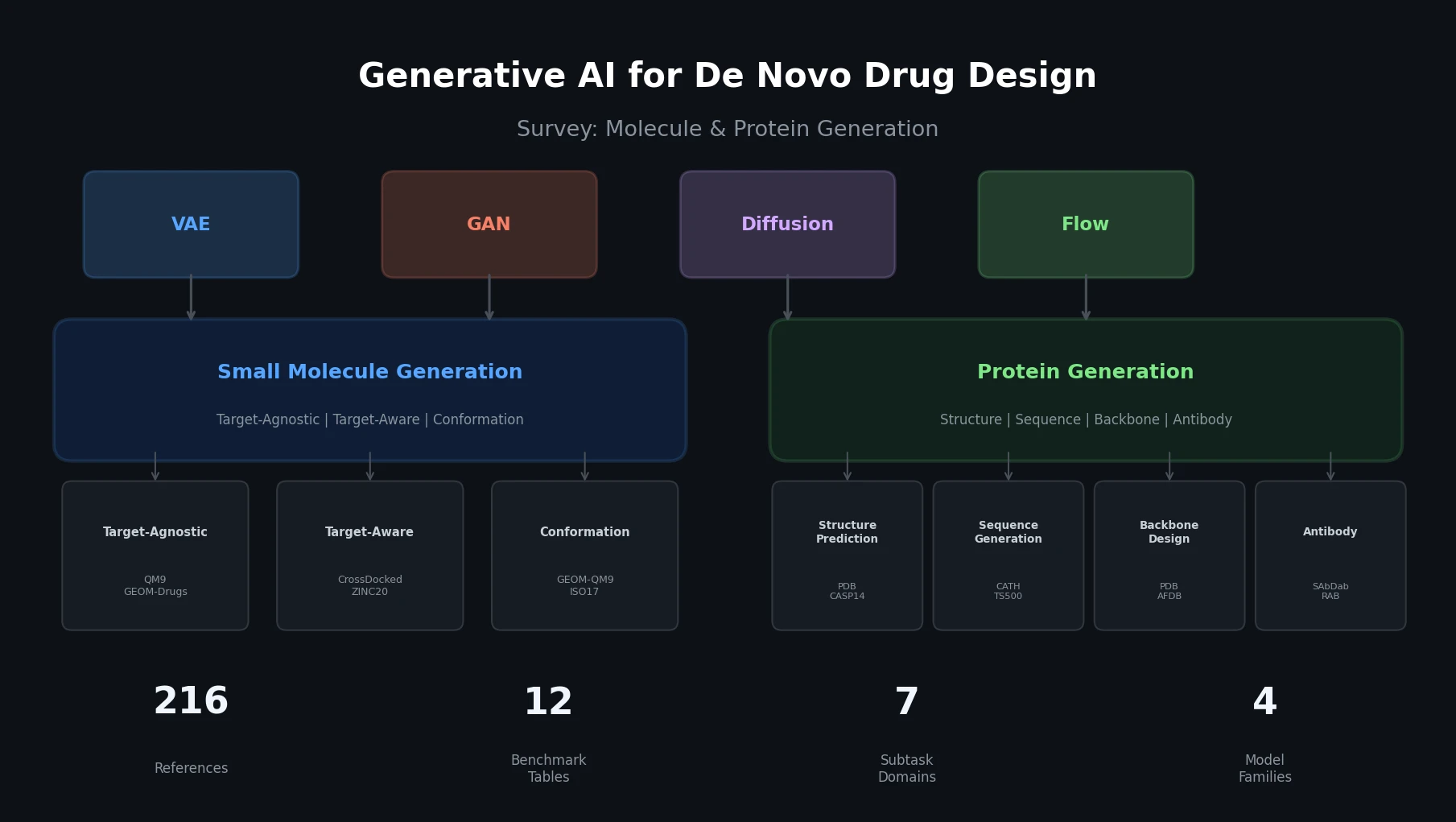
DrugEx v3: Scaffold-Constrained Graph Transformer
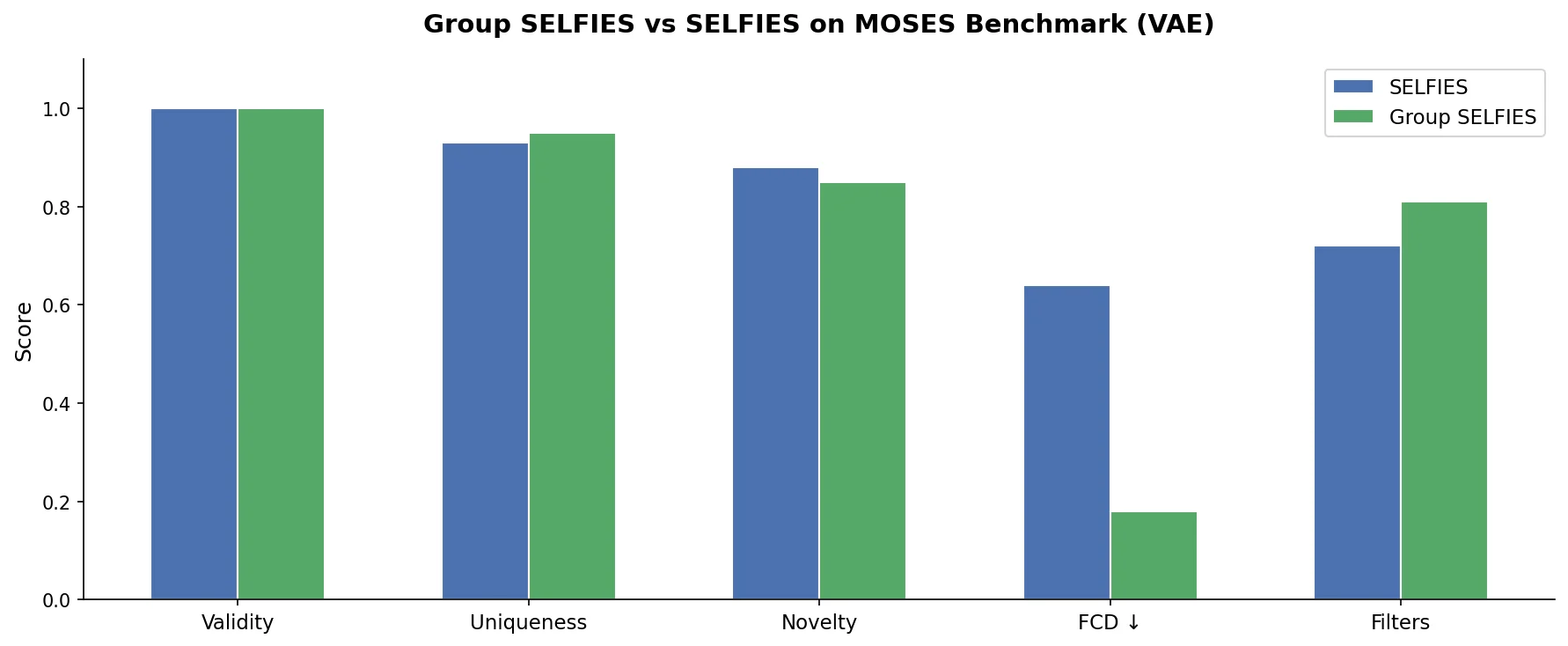
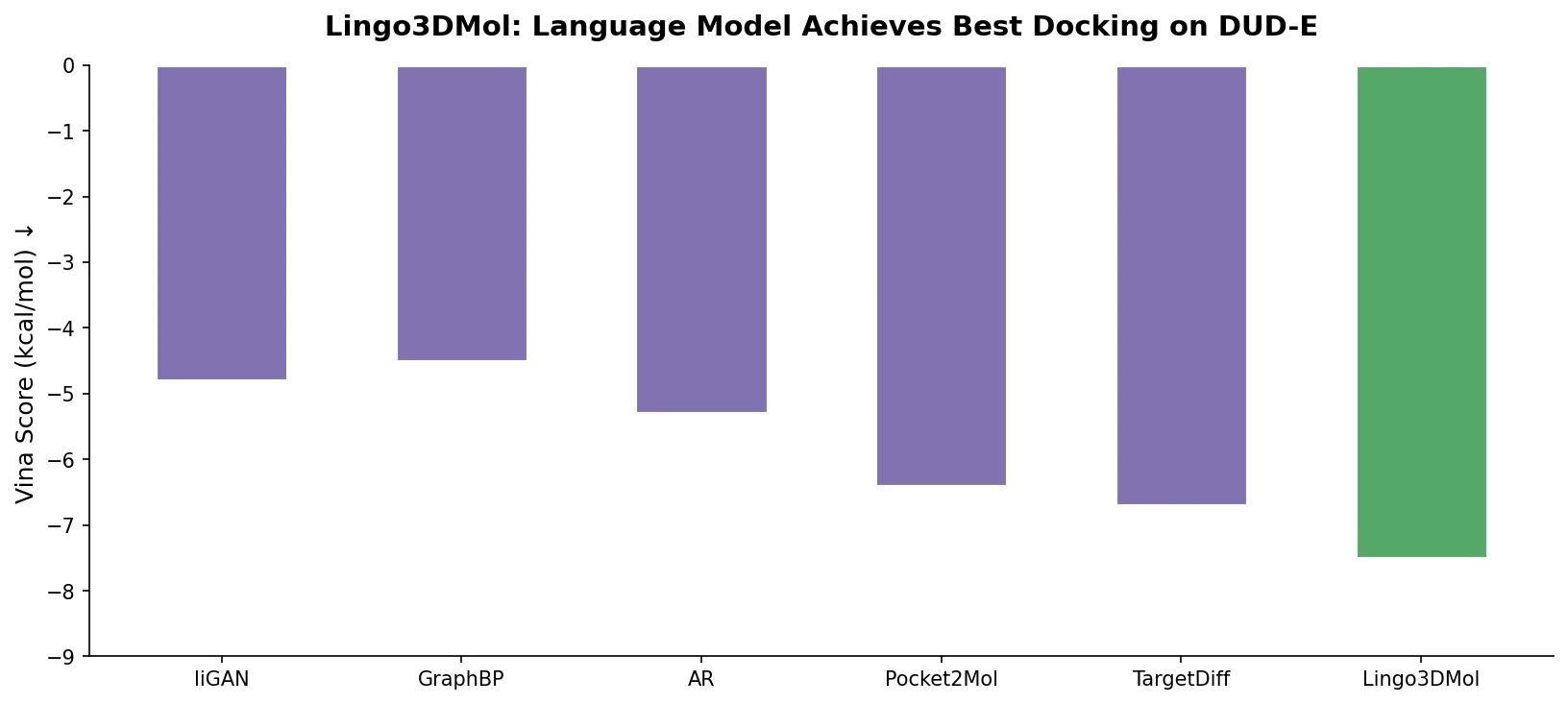
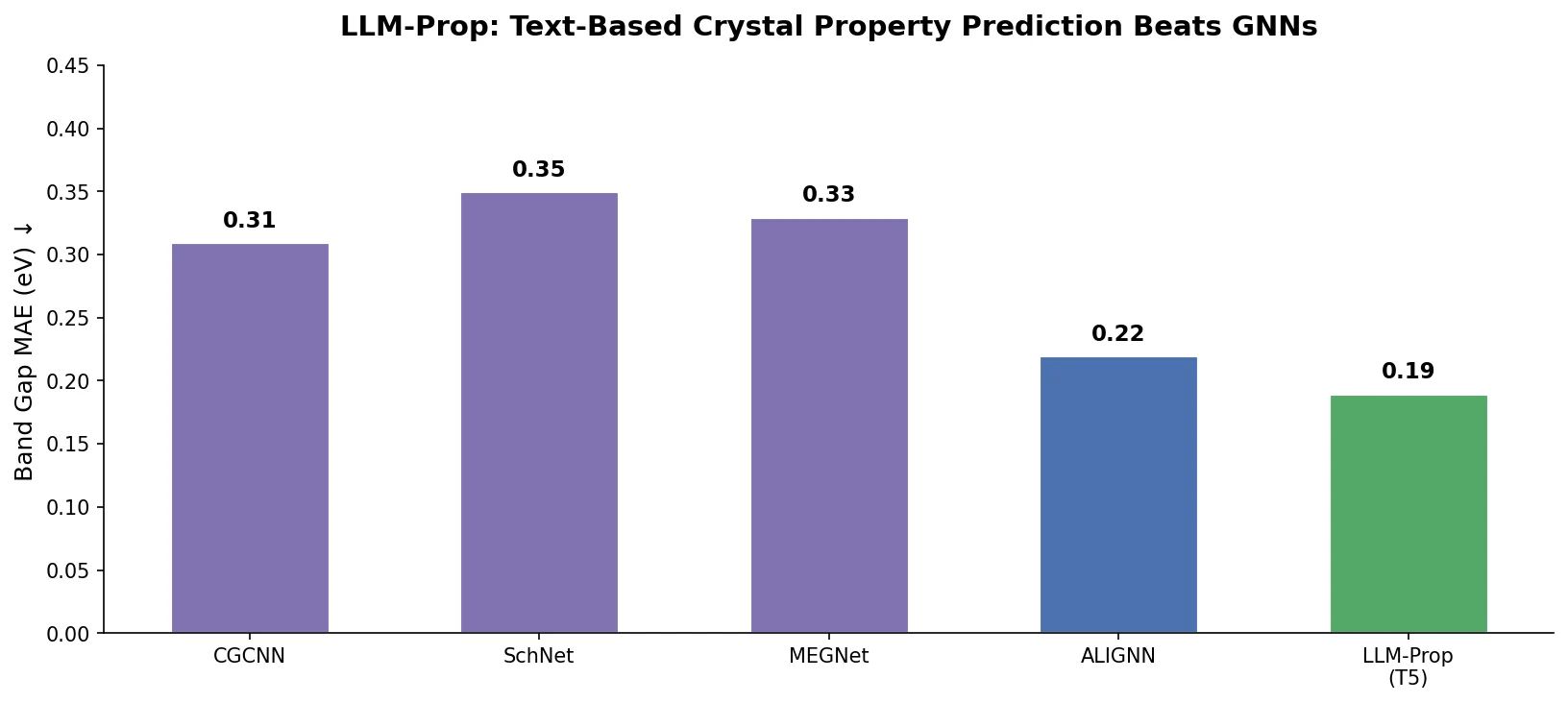
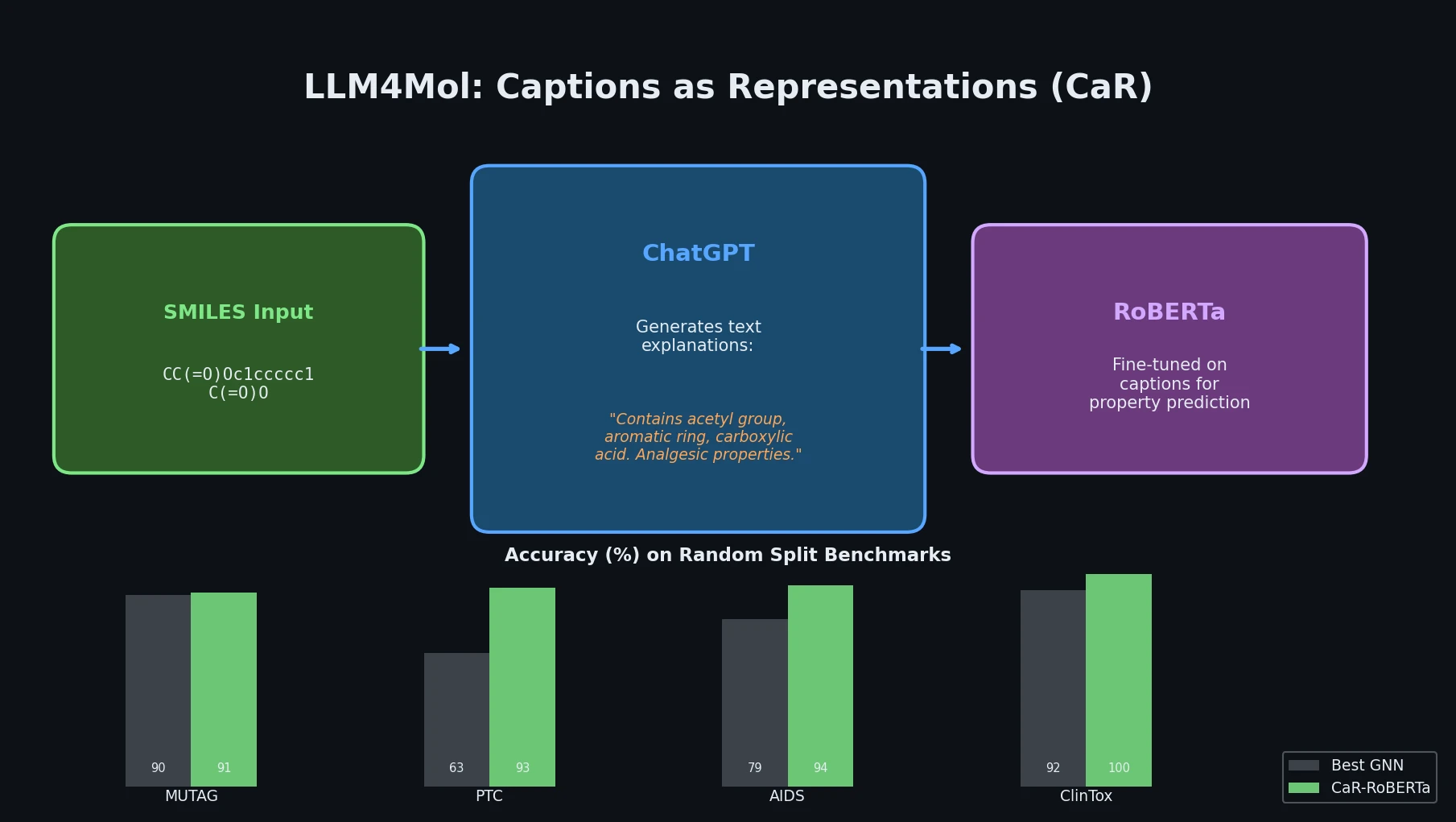
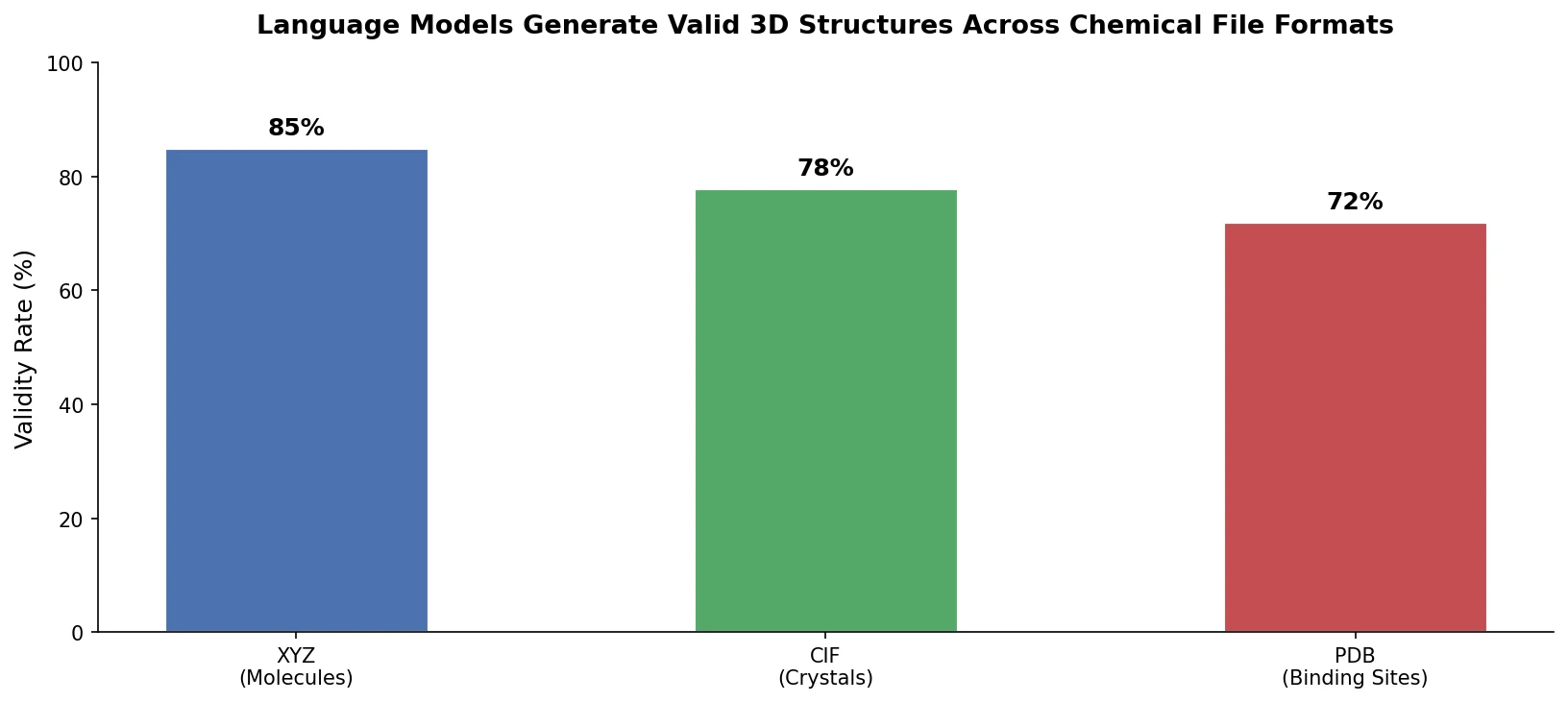
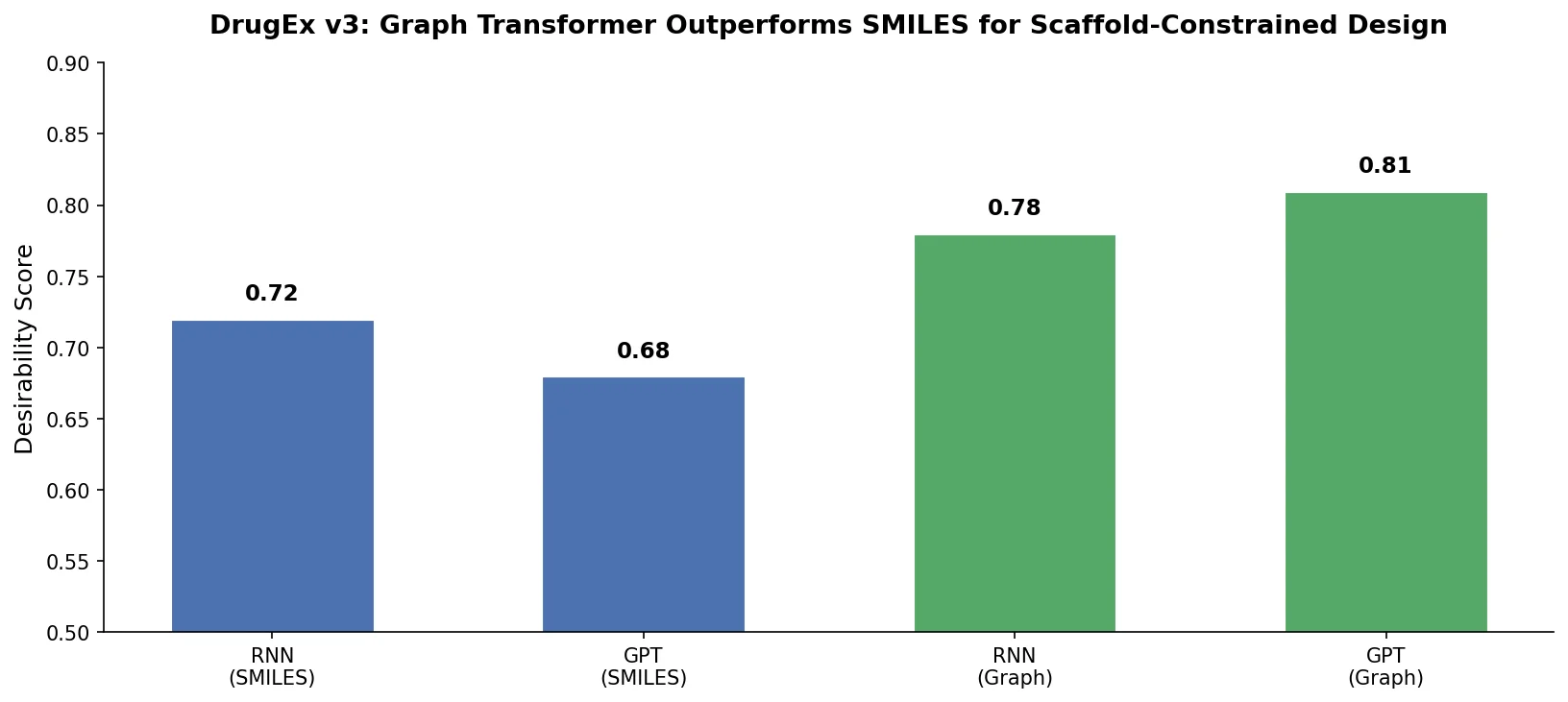
DrugEx v3 extends scaffold-constrained drug design by introducing a Graph Transformer with adjacency-matrix-based positional encoding, achieving 100% molecular validity and high predicted affinity for adenosine A2A receptor ligands.