Embedded-Atom Method: Impurities and Defects in Metals
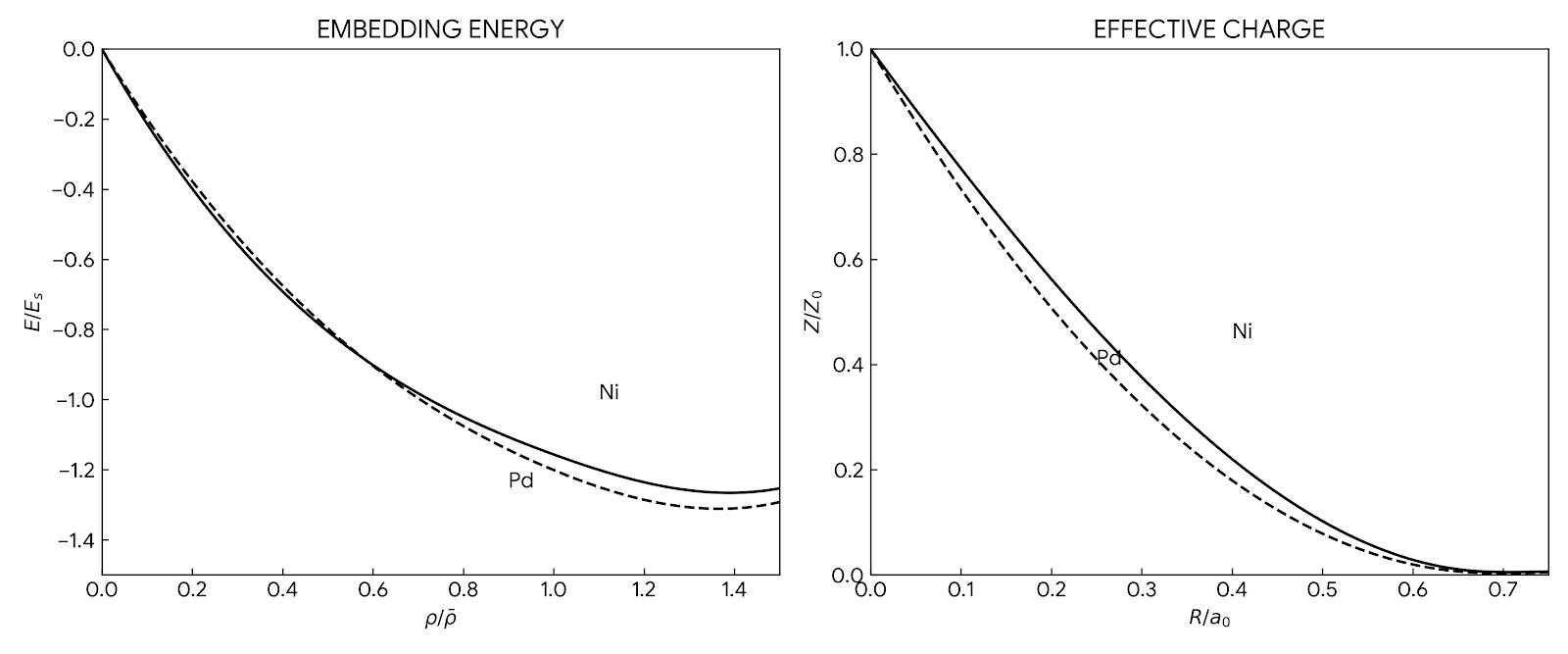
The foundational 1984 paper introducing EAM, a semi-empirical many-body interatomic potential that incorporates density functional theory concepts to accurately simulate metallic systems while maintaining computational efficiency comparable to pair potentials.