MD Simulation of Self-Diffusion on Metal Surfaces (1994)
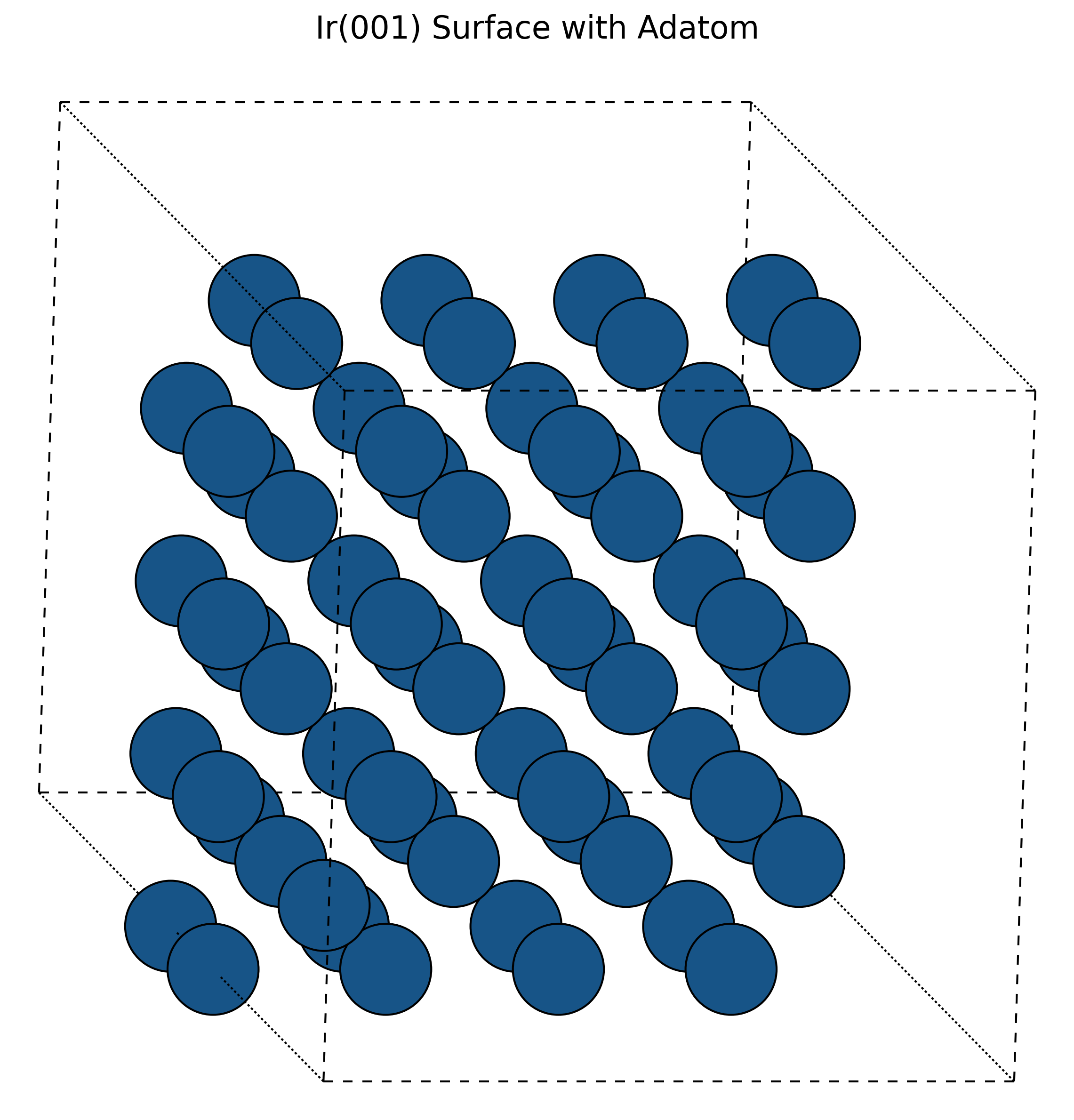
A molecular dynamics investigation using EAM and many-body potentials to elucidate atomic exchange mechanisms on Iridium surfaces, verifying Field Ion Microscope observations.
A molecular dynamics investigation using EAM and many-body potentials to elucidate atomic exchange mechanisms on Iridium surfaces, verifying Field Ion Microscope observations.
A comprehensive categorization of OCSR methods, organizing techniques by their fundamental approach: deep learning, traditional ML, and rule-based systems.
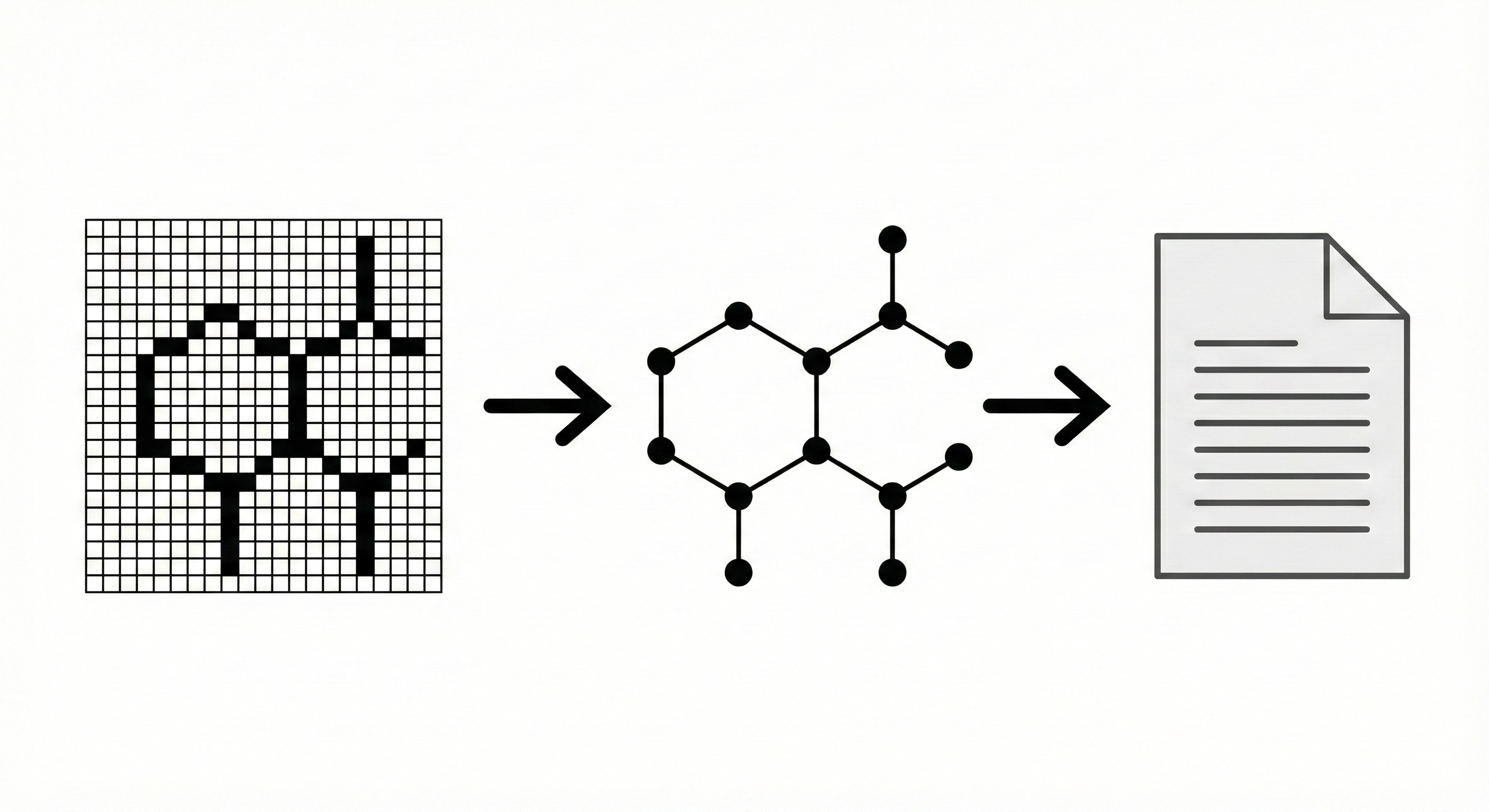
This paper describes an early prototype system that digitizes chemical structure diagrams from scanned documents. It employs a multi-stage pipeline involving convex bounding polygon extraction, vectorization, and rule-based heuristics to generate MDL Molfiles.
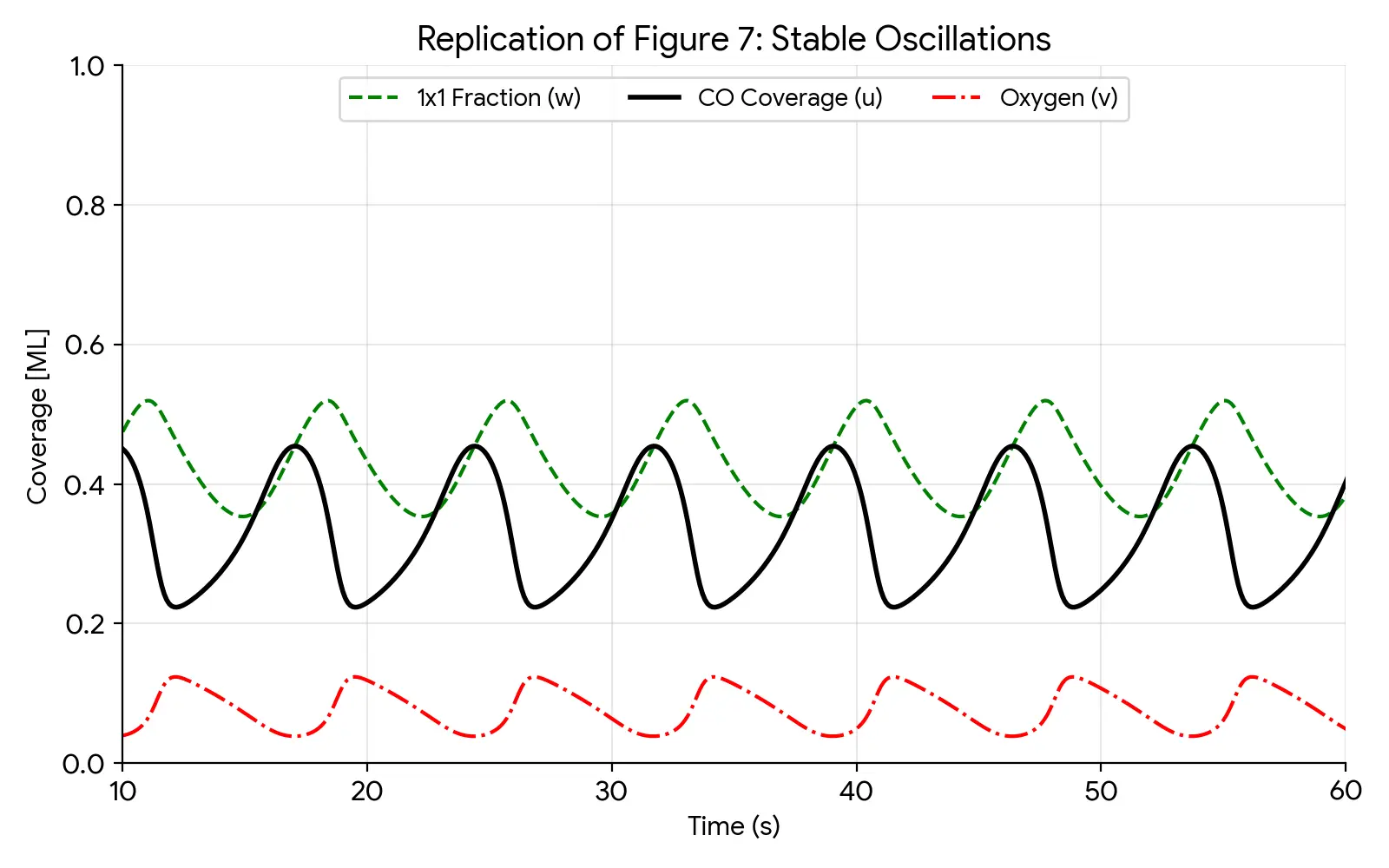
This paper presents a 4-variable kinetic model coupling surface reaction dynamics with structural phase transitions to reproduce complex oscillatory behavior on Pt(110).
This paper presents OSRA, the first open-source utility for converting graphical chemical structures from documents into machine-readable formats (SMILES/SD). It outlines a pipeline combining existing image processing tools with custom heuristics for bond and atom detection, establishing a foundation for accessible chemical information extraction.
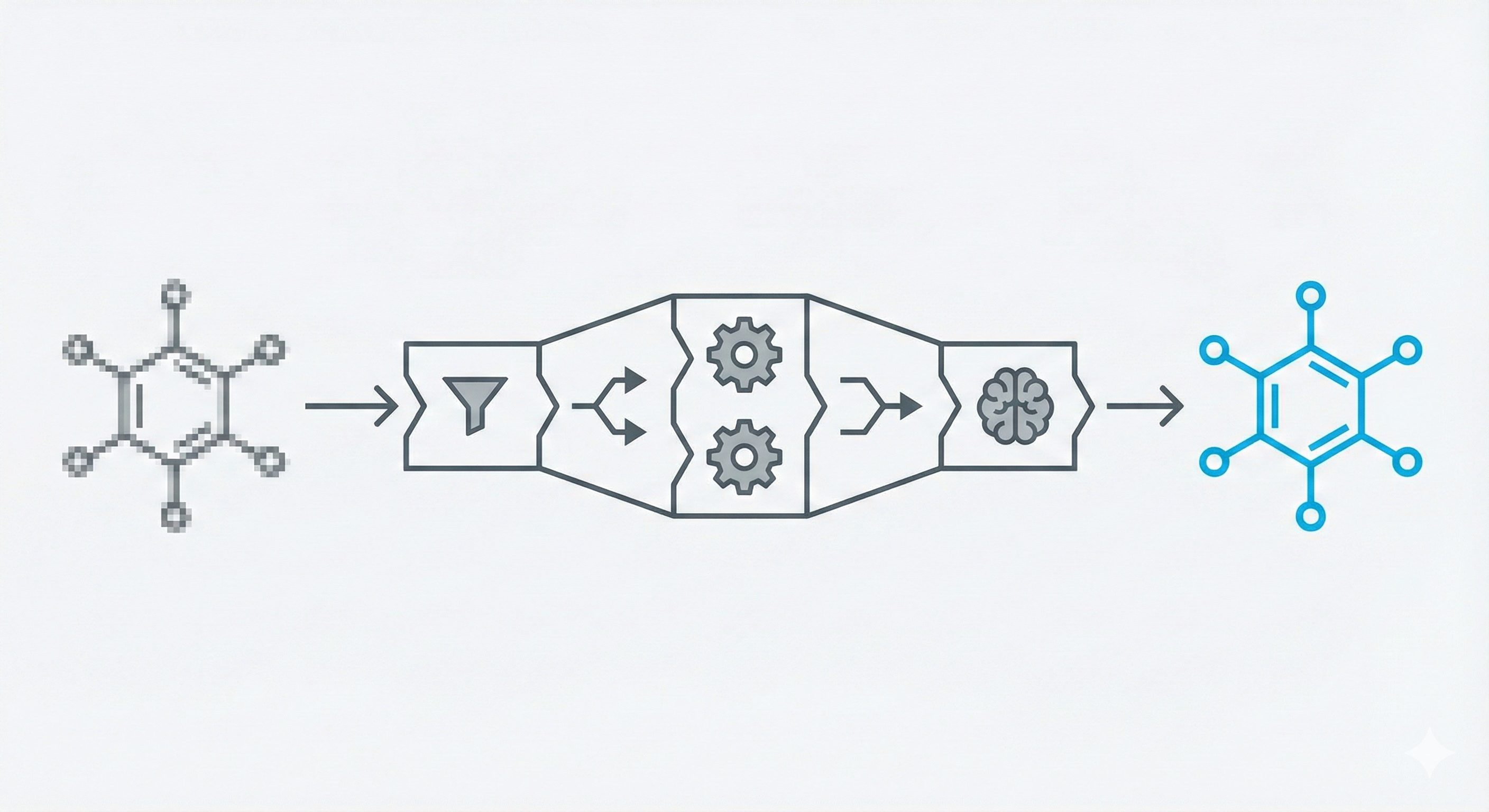
This methodological paper proposes a comprehensive pipeline to digitize chemical structure images. It achieves 97% reconstruction accuracy on benchmarks by combining a topology-preserving vectorizer with a chemical knowledge validation module.
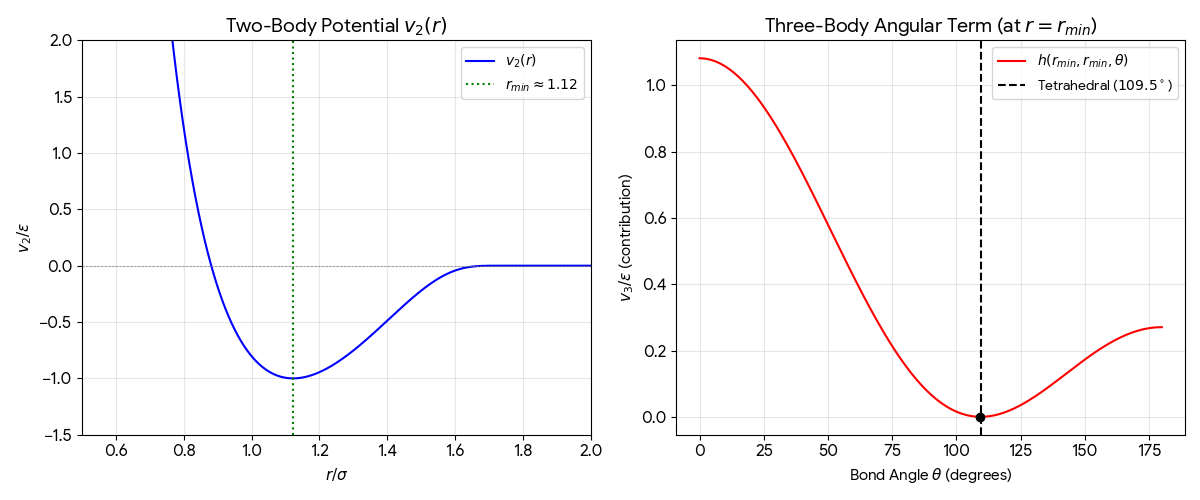
Stillinger and Weber propose a 3-body interaction potential that stabilizes the diamond crystal structure of silicon and reproduces liquid properties through molecular dynamics, addressing the inability of standard pair potentials to model tetrahedral semiconductors.
This molecular dynamics study reveals that adatom dimers on fcc(111) surfaces exhibit simultaneous multiple jumps at intermediate temperatures, migrating with mobility comparable to single adatoms.
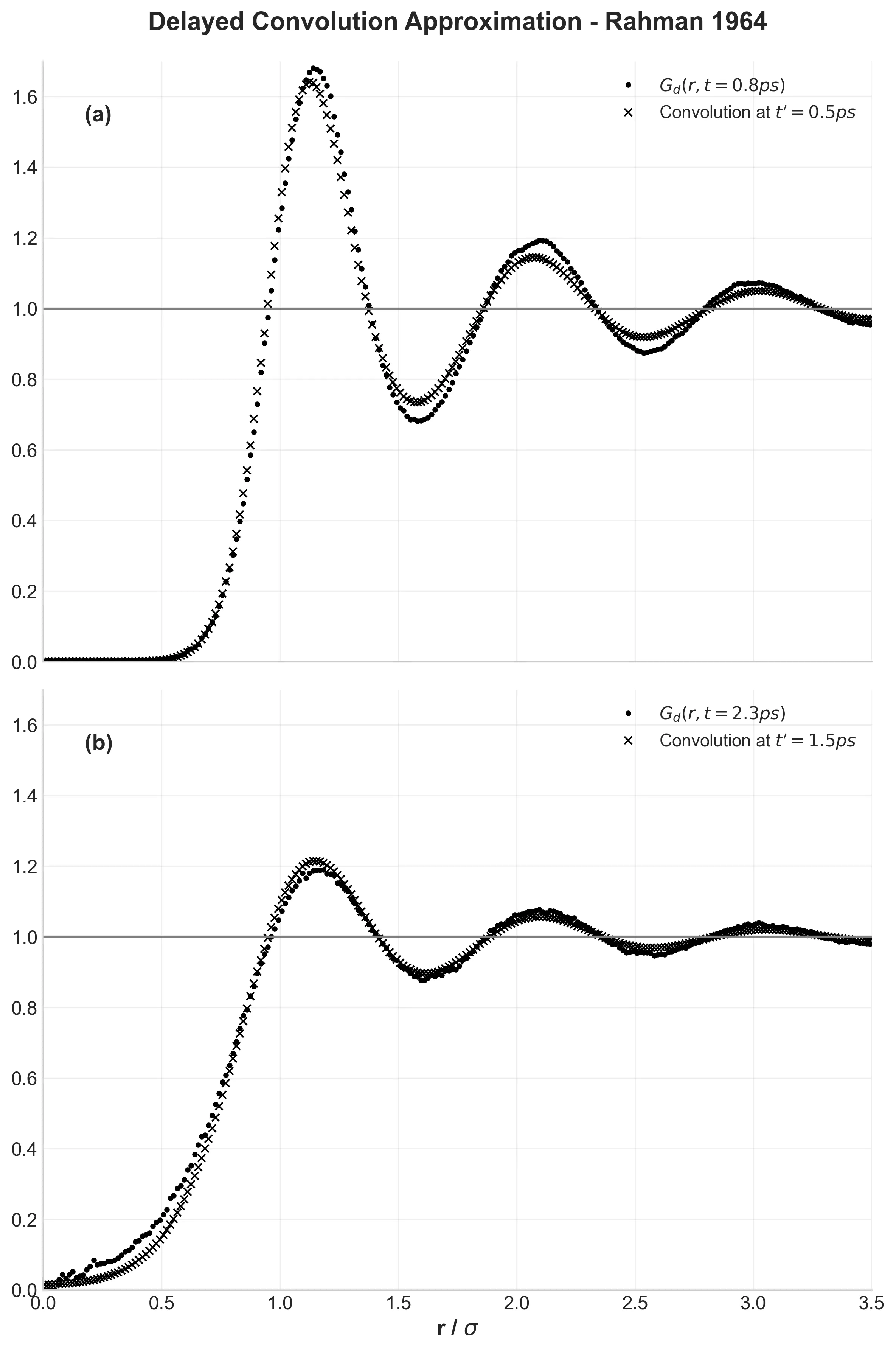
This work validated classical Molecular Dynamics for simulating liquids, revealing the ‘cage effect’ in velocity autocorrelation and establishing predictor-corrector integration algorithms for N-body problems.
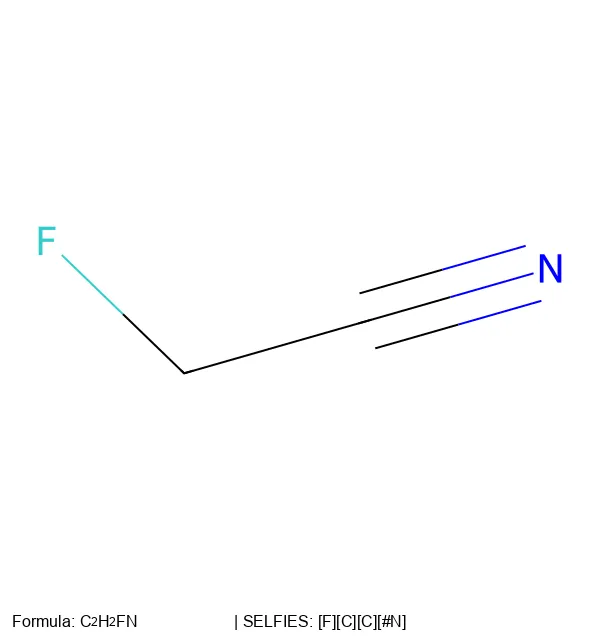
A 2024 Nature Machine Intelligence paper providing causal evidence that invalid SMILES generation improves chemical language model performance by filtering low-likelihood samples, while validity constraints (as in SELFIES) introduce structural biases that impair distribution learning.
This 2022 perspective paper reviews 250 years of chemical notation evolution and proposes 16 concrete research projects to extend SELFIES beyond traditional organic chemistry into polymers, crystals, and reactions.
A comprehensive 2020 analysis of the tautomerism problem in chemical databases, compiling 86 tautomeric transformation rules (20 existing, 66 new) and validating them across 400M+ structures to inform algorithmic improvements for InChI V2.