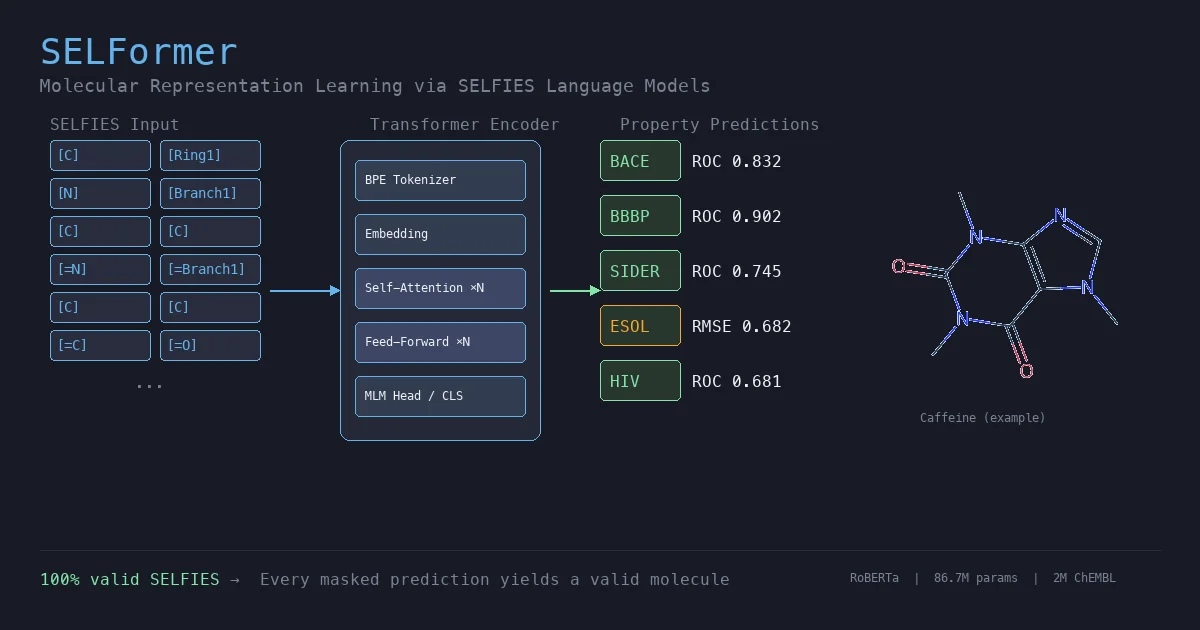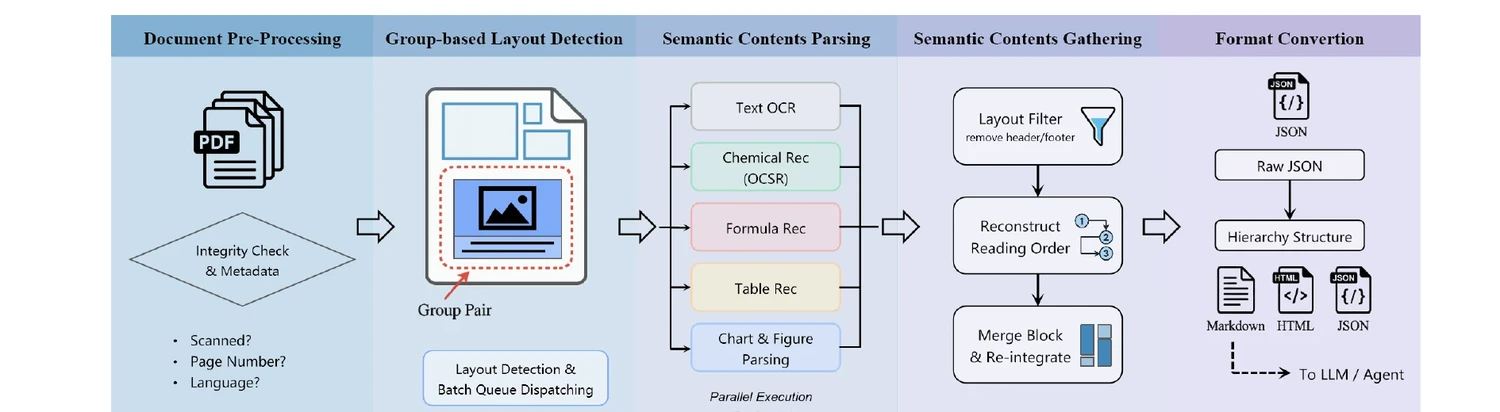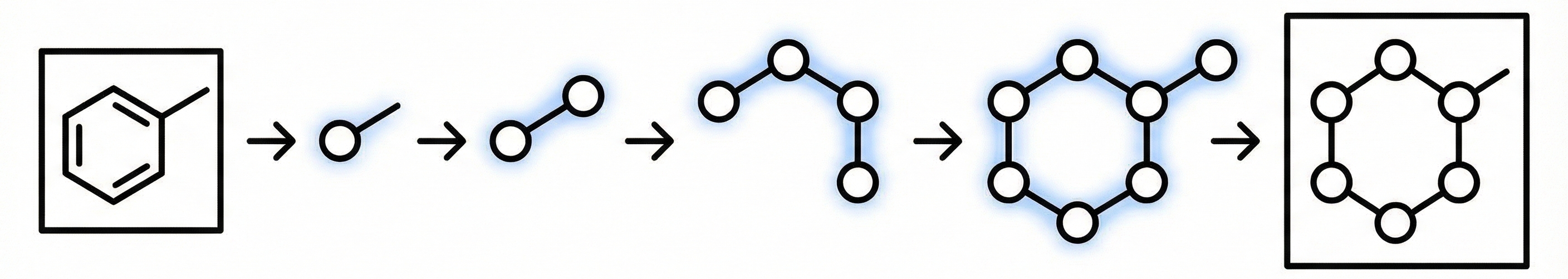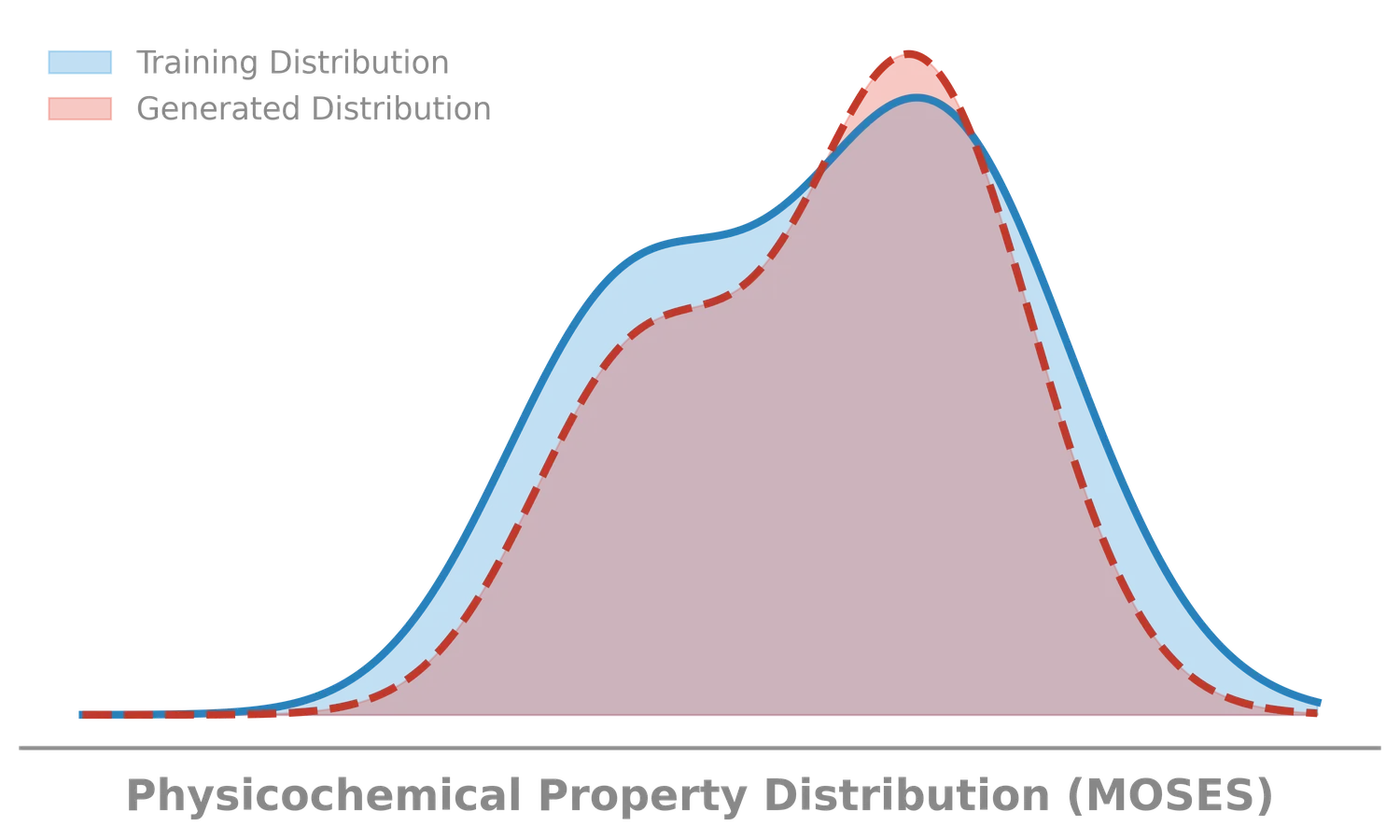
MoLFormer: Large-Scale Chemical Language Representations
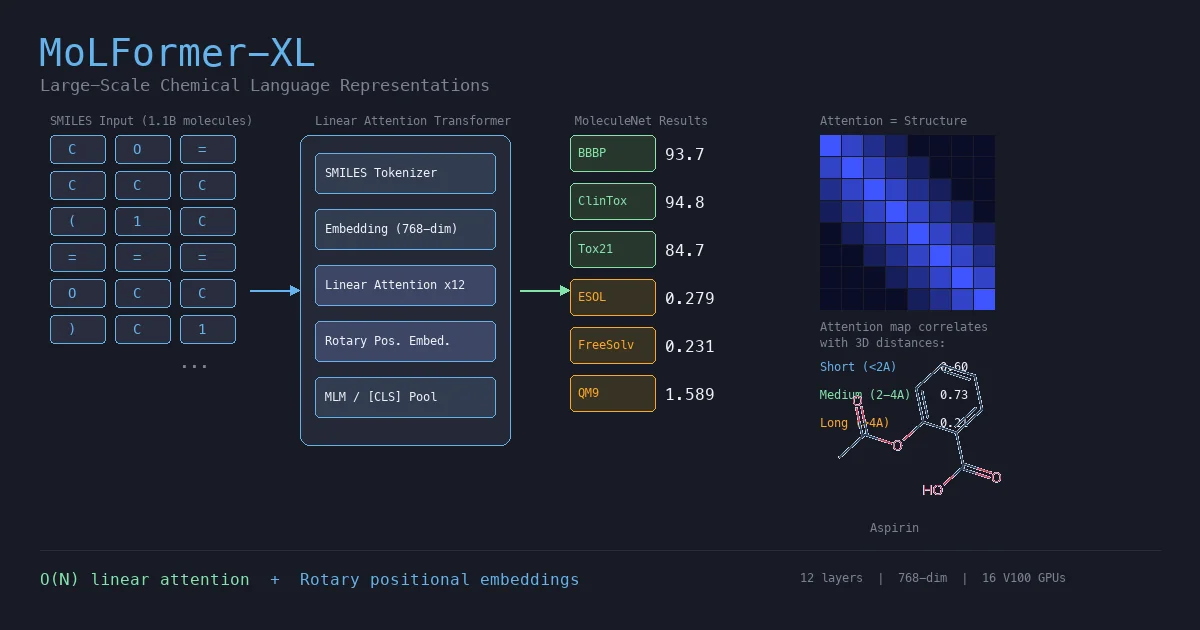
MoLFormer is a transformer encoder with linear attention and rotary positional embeddings, pretrained via masked language modeling on 1.1 billion molecules from PubChem and ZINC. MoLFormer-XL outperforms GNN baselines on most MoleculeNet classification and regression tasks, and attention analysis reveals that the model learns interatomic spatial relationships directly from SMILES strings.