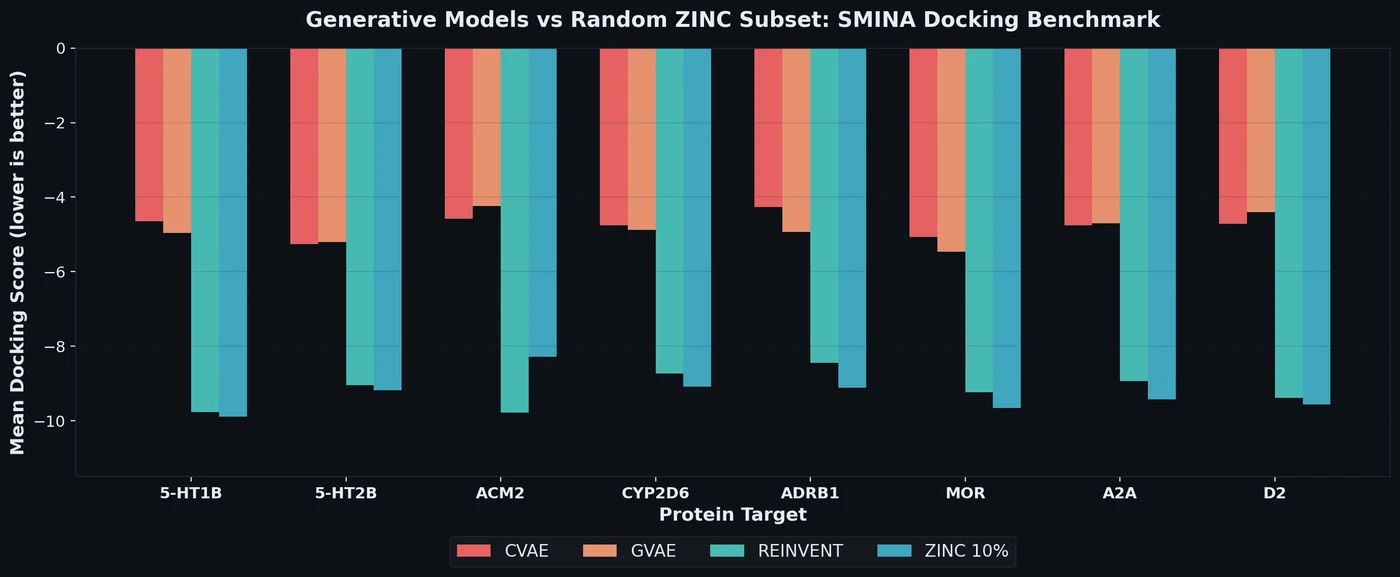
SMINA Docking Benchmark for De Novo Drug Design Models
Proposes a benchmark for de novo drug design using SMINA docking scores across eight drug targets, revealing that popular generative models fail to outperform random ZINC subsets.
Proposes a benchmark for de novo drug design using SMINA docking scores across eight drug targets, revealing that popular generative models fail to outperform random ZINC subsets.
Tartarus introduces a modular suite of realistic molecular design benchmarks grounded in computational chemistry simulations. Benchmarking eight generative models reveals that no single algorithm dominates all tasks, and simple genetic algorithms often outperform deep generative models.
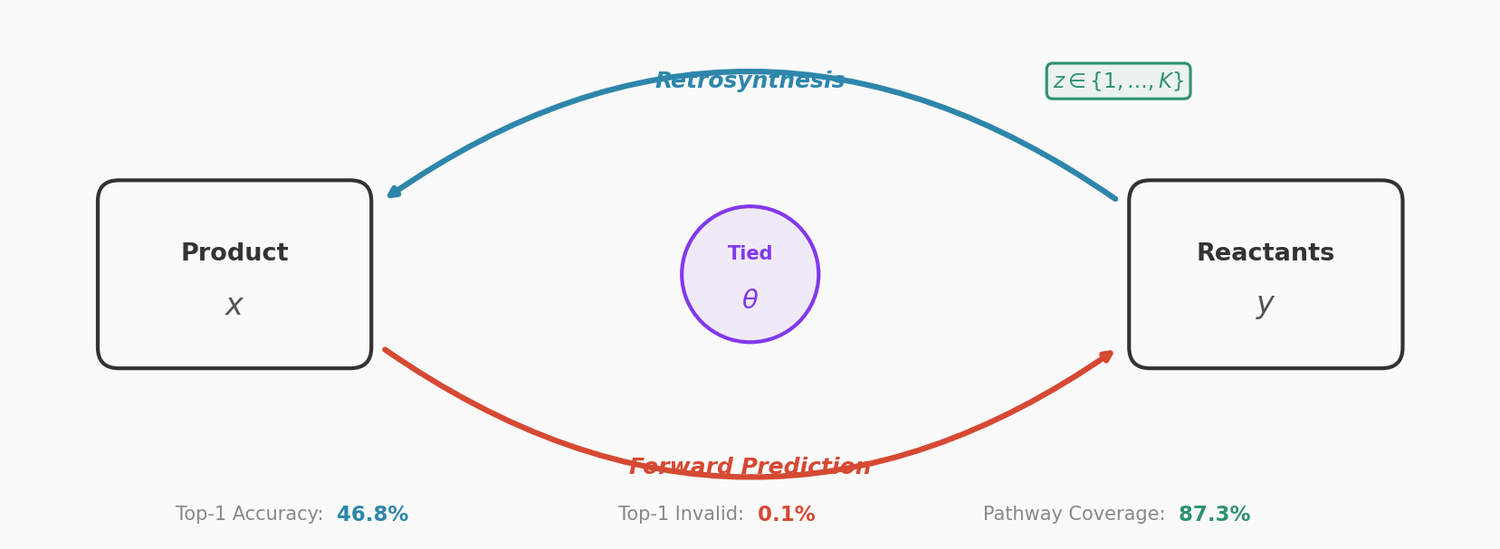
This paper couples a retrosynthesis transformer with a forward reaction transformer through parameter sharing, cycle consistency checks, and multinomial latent variables. The combined approach reduces top-1 SMILES invalidity to 0.1% on USPTO-50K, improves top-10 accuracy to 78.5%, and achieves 87.3% pathway coverage on a multi-pathway in-house dataset.
BARTSmiles pre-trains a BART-large model on 1.7 billion SMILES strings from ZINC20 and achieves the best reported results on 11 classification, regression, and generation benchmarks.
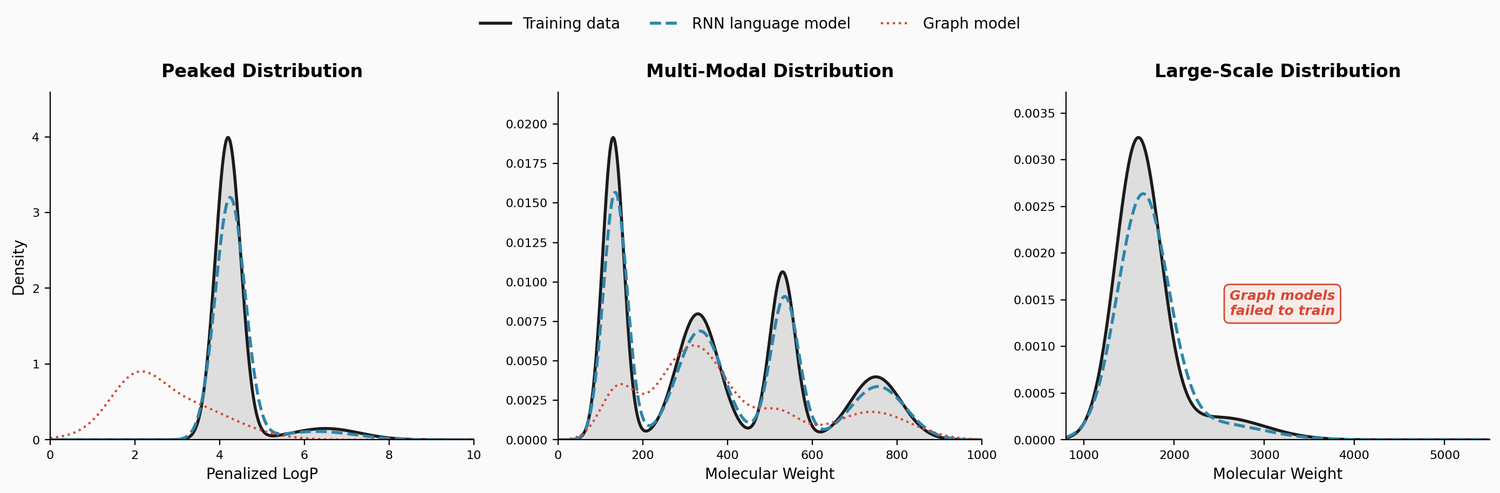
This study benchmarks RNN-based chemical language models against graph generative models on three challenging tasks: high penalized LogP distributions, multi-modal molecular distributions, and large-molecule generation from PubChem. The LSTM language models consistently outperform JTVAE and CGVAE.
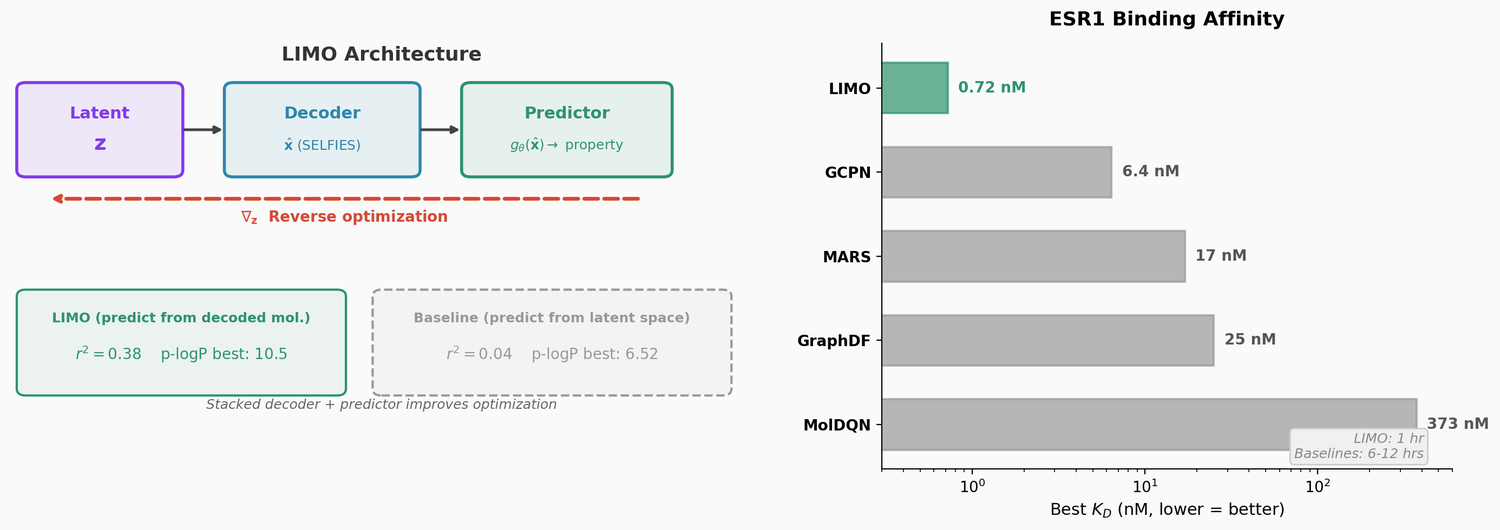
LIMO combines a SELFIES-based VAE with a novel stacked property predictor architecture (decoder output as predictor input) and gradient-based reverse optimization on the latent space. It is 6-8x faster than RL baselines and 12x faster than sampling methods while generating molecules with nanomolar binding affinities, including a predicted KD of 6e-14 M against the human estrogen receptor.
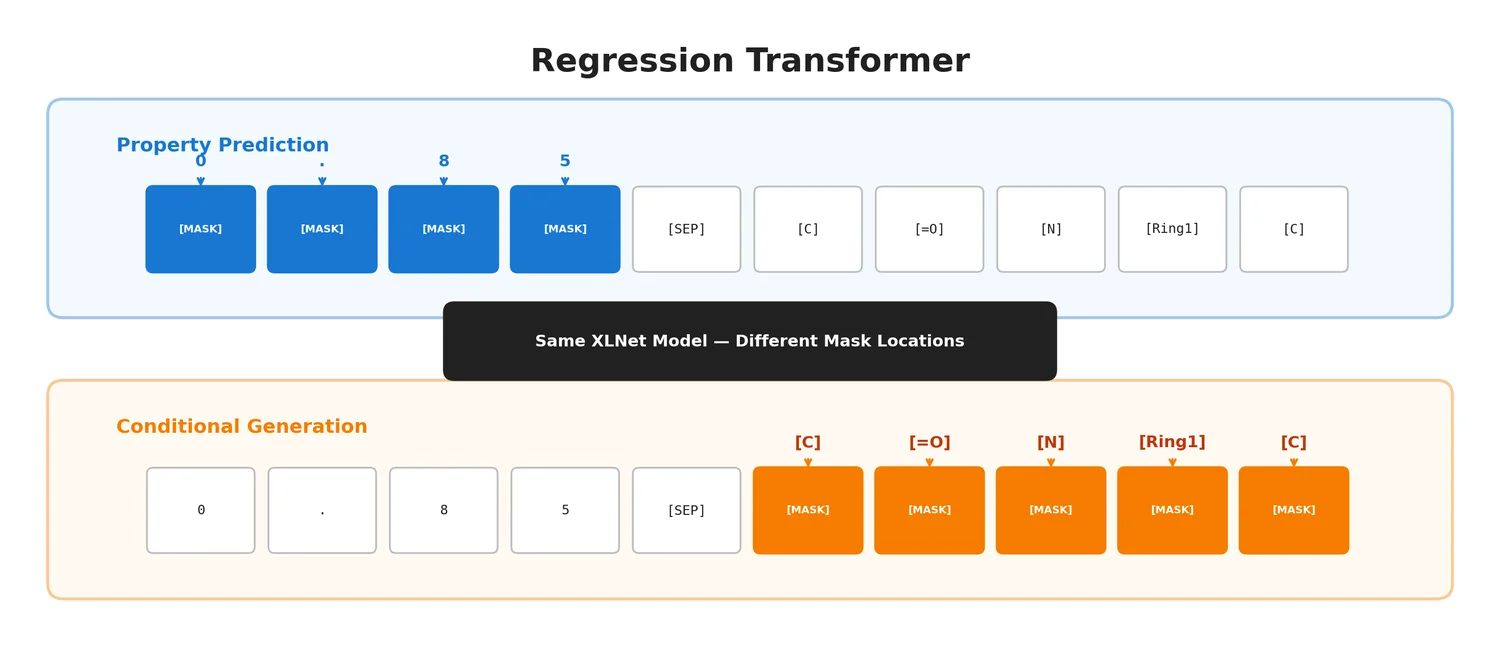
The Regression Transformer (RT) reformulates regression as conditional sequence modelling, enabling a single XLNet-based model to both predict continuous molecular properties and generate novel molecules conditioned on desired property values.
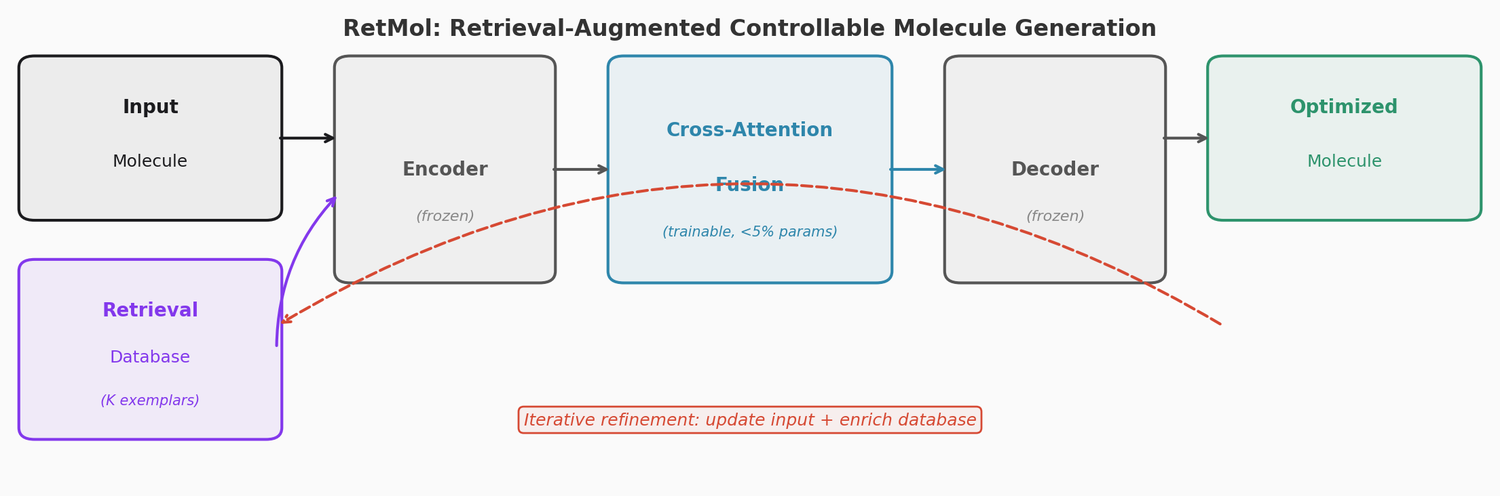
RetMol plugs a lightweight cross-attention retrieval module into a pre-trained Chemformer backbone to guide molecule generation toward multi-property design criteria. It requires no task-specific fine-tuning and works with as few as 23 exemplar molecules. It achieves 94.5% success on QED optimization, 96.9% on GSK3b/JNK3 dual inhibitor design, and 2.84 kcal/mol average binding affinity improvement on SARS-CoV-2 main protease inhibitor optimization.
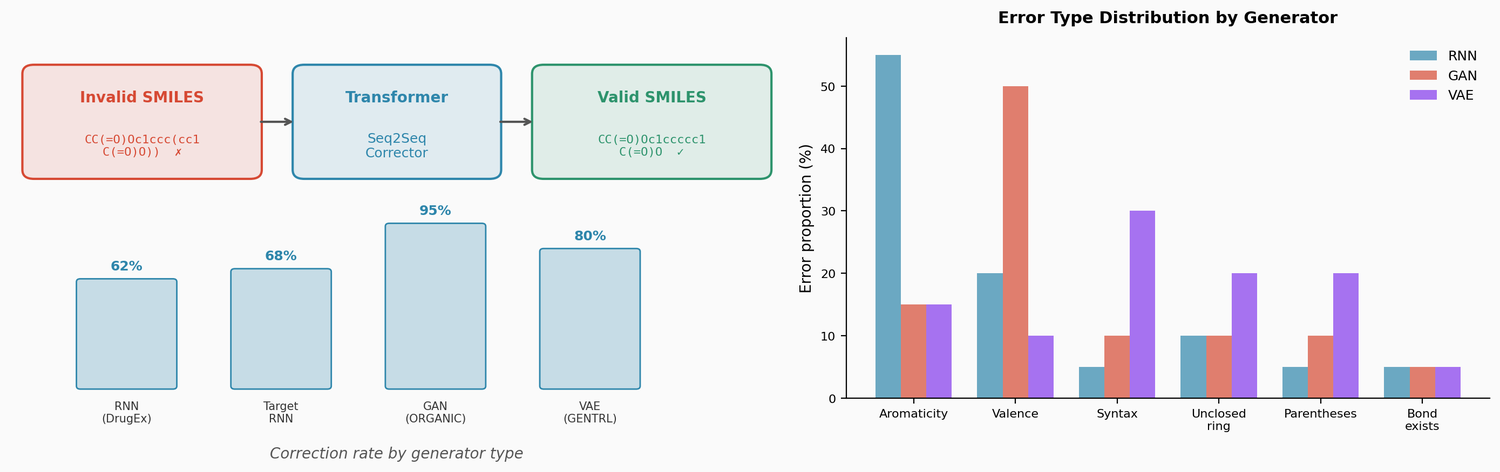
This paper trains a transformer model to correct invalid SMILES produced by de novo molecular generators (RNN, VAE, GAN). The corrector fixes 60-95% of invalid outputs, and the fixed molecules are comparable in novelty and similarity to valid generator outputs. The approach also enables local chemical space exploration by introducing and correcting errors in existing molecules.
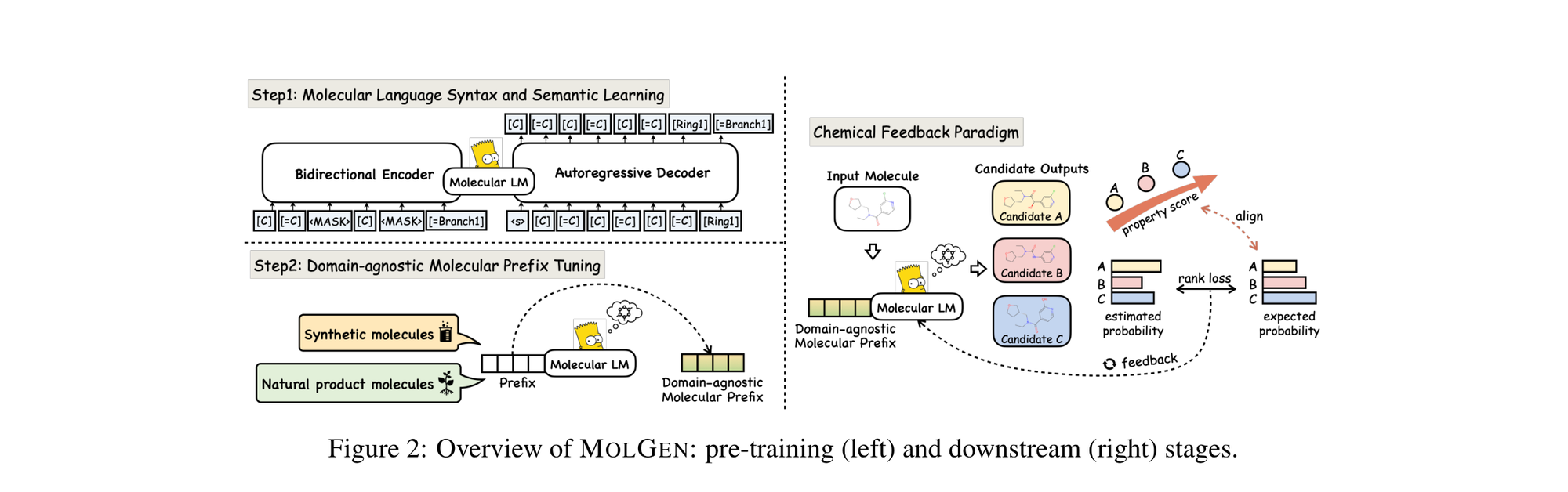
MolGen pre-trains on 100M+ SELFIES molecules, introduces domain-agnostic prefix tuning for cross-domain transfer, and applies a chemical feedback paradigm to reduce molecular hallucinations.
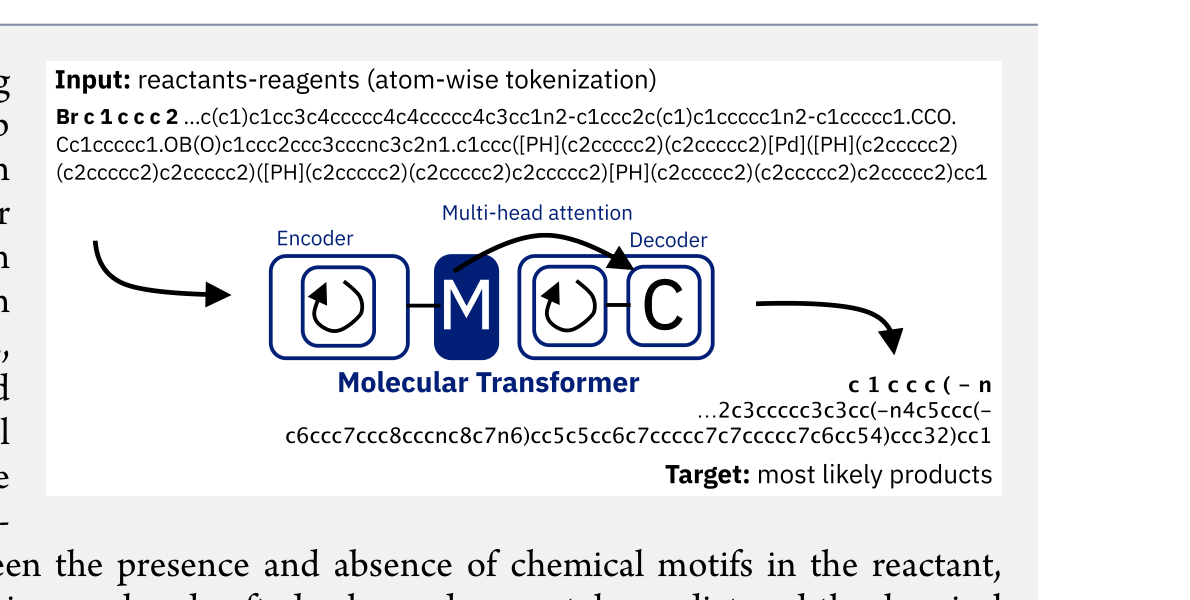
The Molecular Transformer applies the Transformer architecture to forward reaction prediction, treating it as SMILES-to-SMILES machine translation. It achieves 90.4% top-1 accuracy on USPTO_MIT, outperforms quantum-chemistry baselines on regioselectivity, and provides calibrated uncertainty scores (0.89 AUC-ROC) for ranking synthesis pathways.
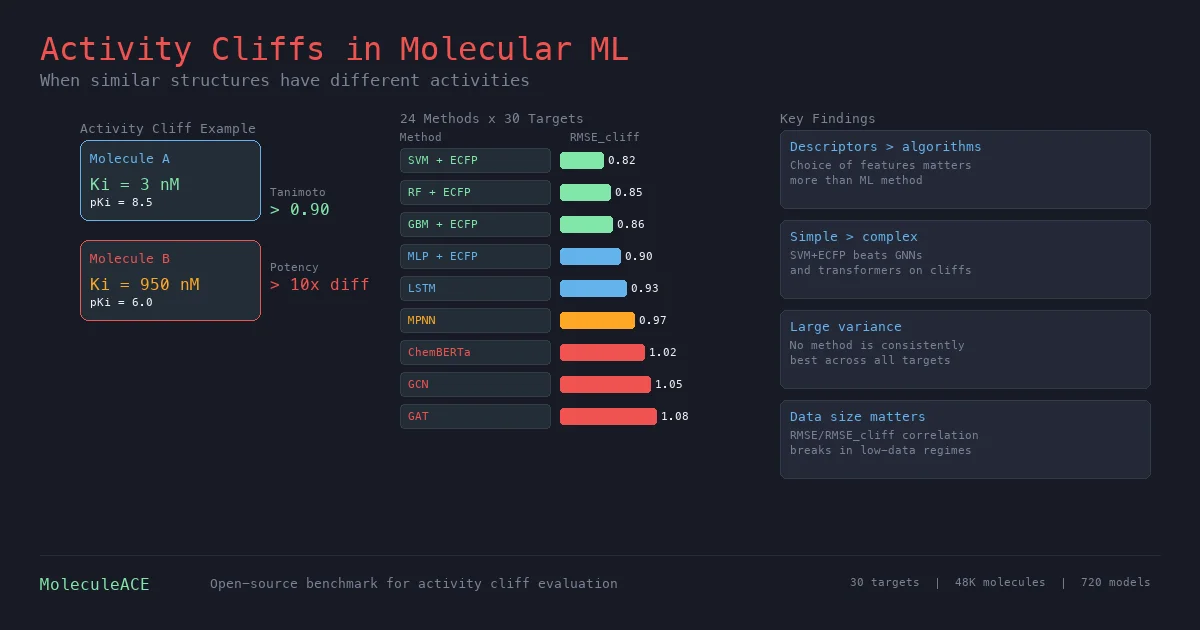
This paper benchmarks 24 machine and deep learning methods on activity cliff compounds (structurally similar molecules with large potency differences) across 30 macromolecular targets. Traditional ML with molecular fingerprints consistently outperforms graph neural networks and SMILES-based transformers on these challenging cases, especially in low-data regimes.