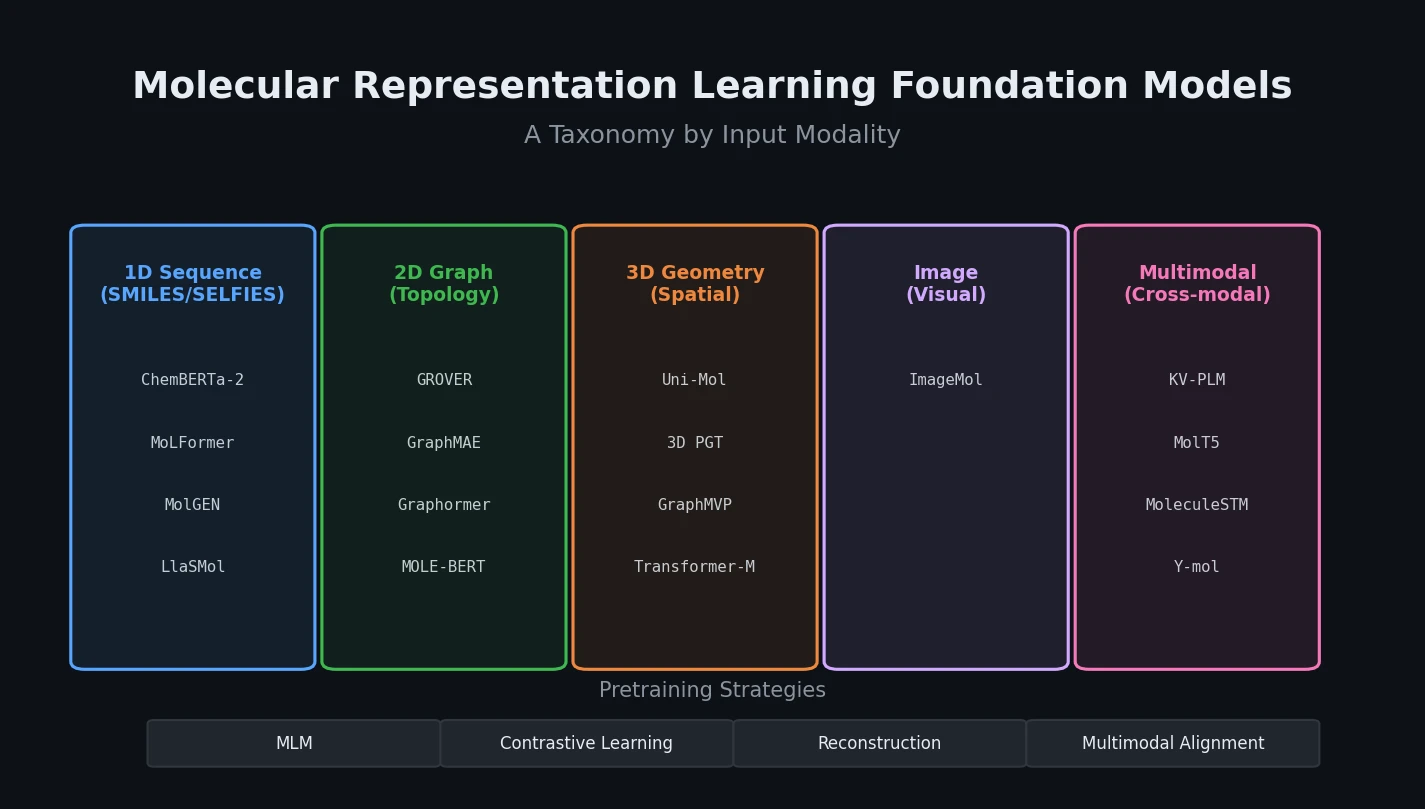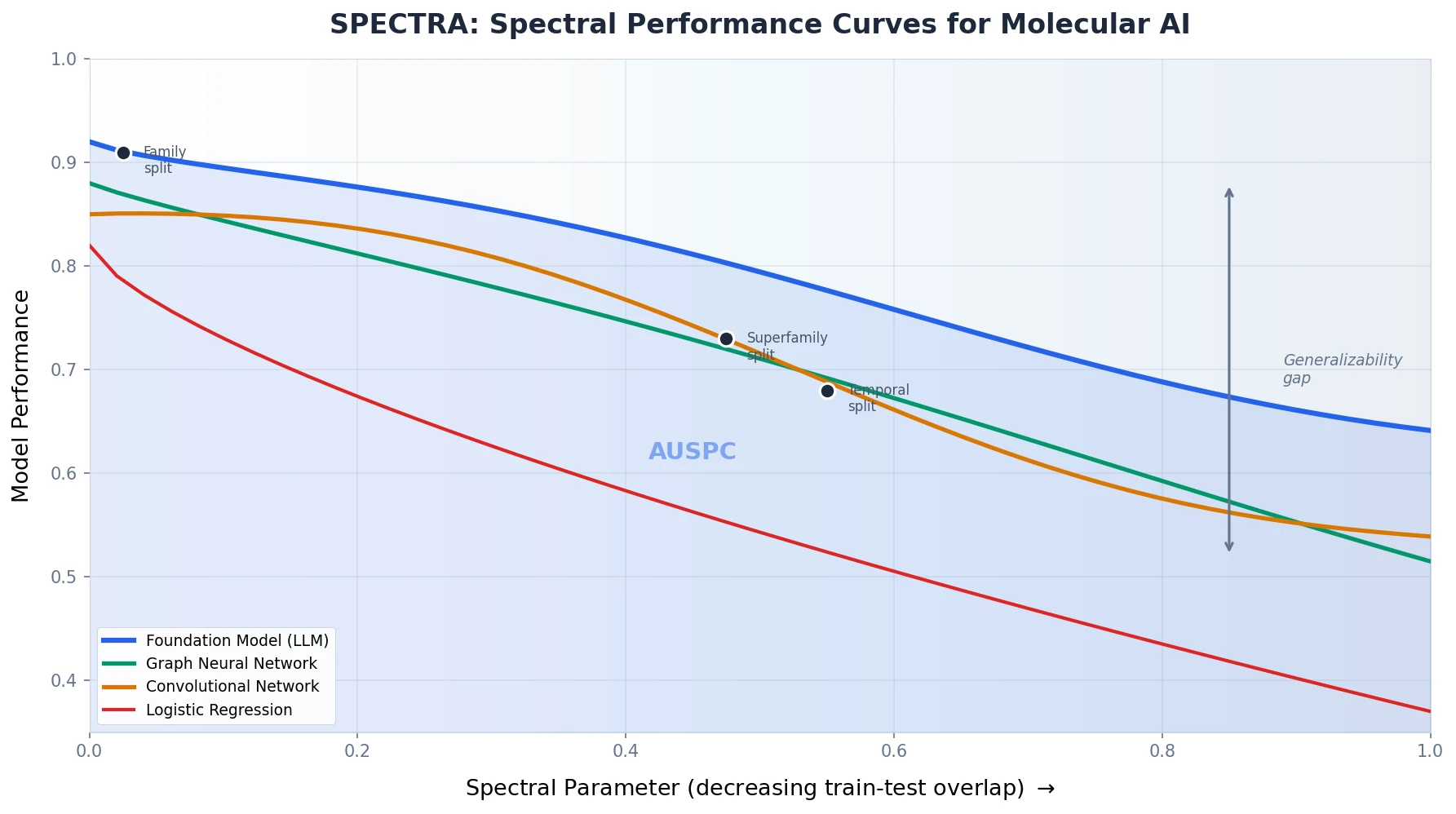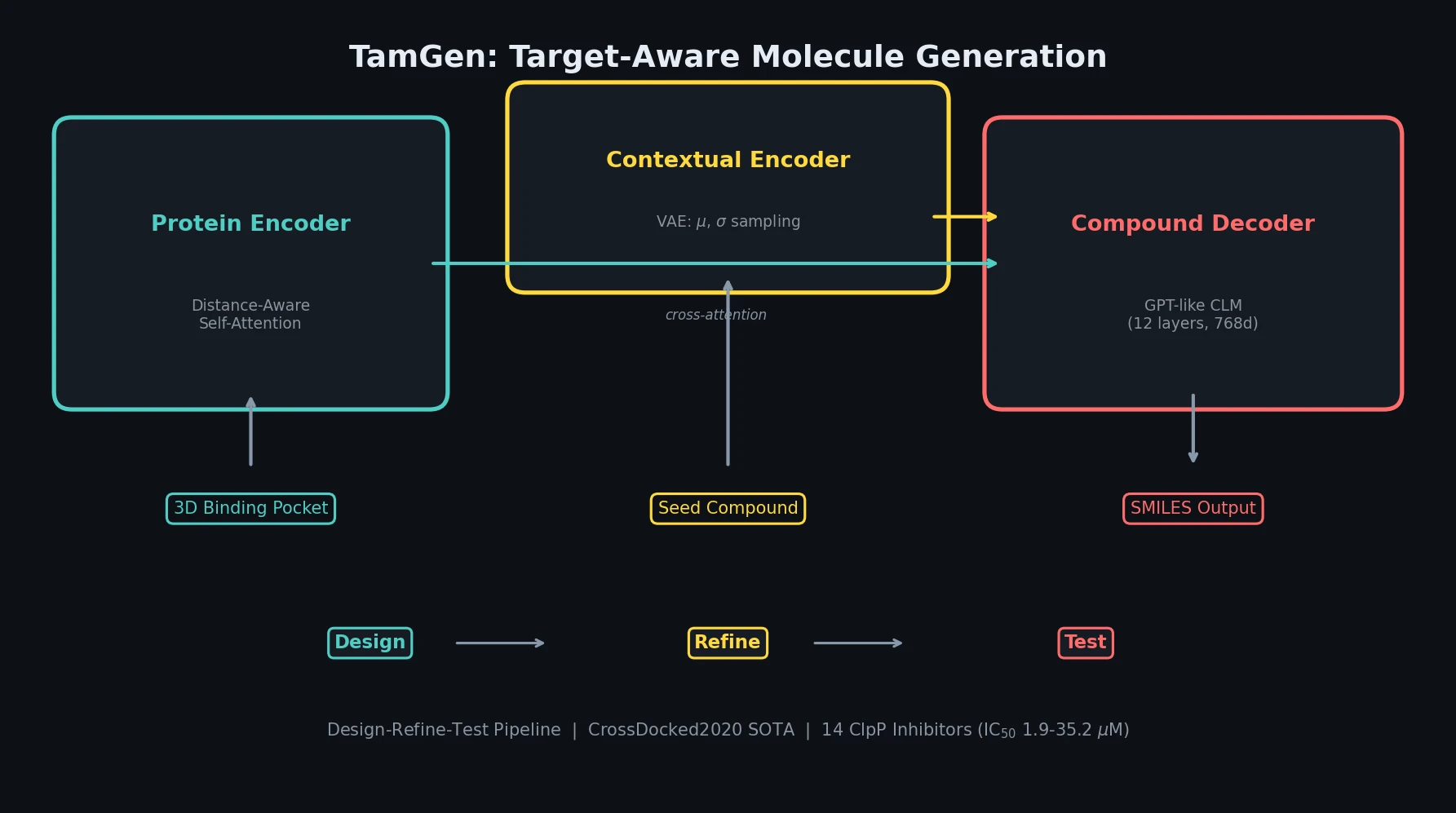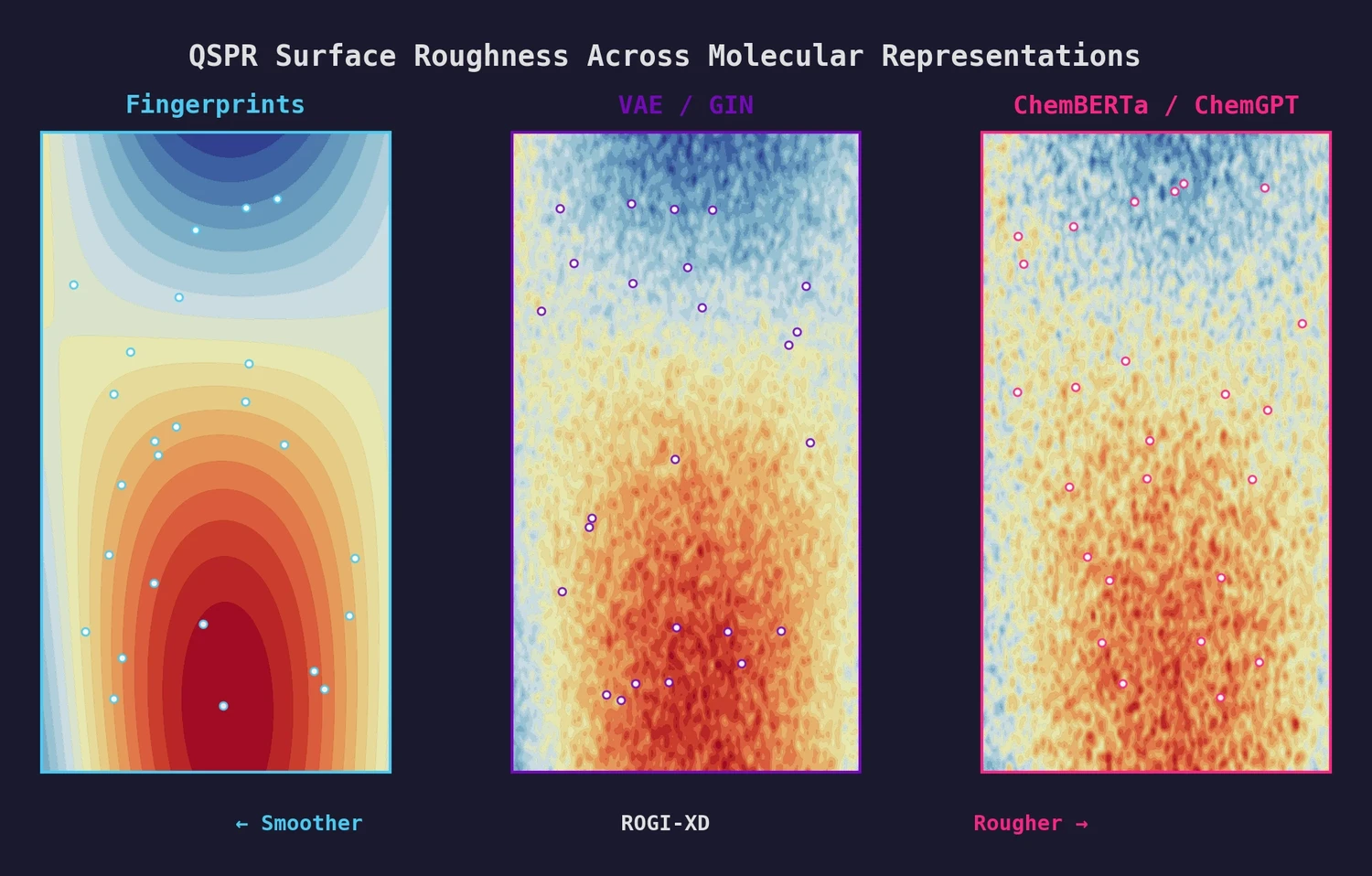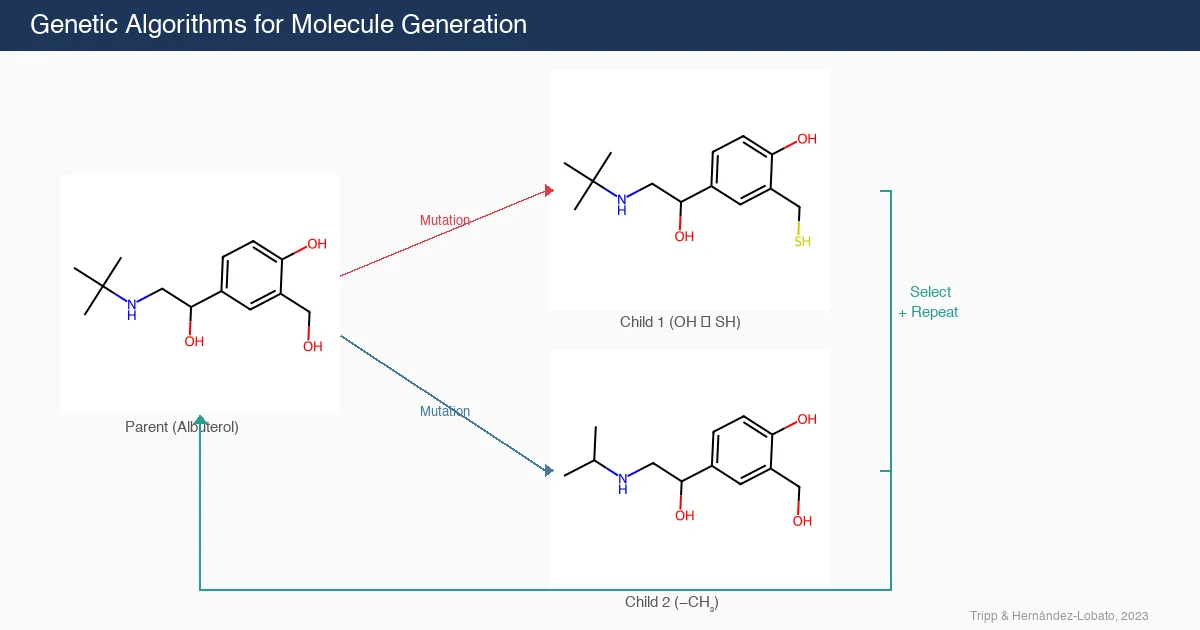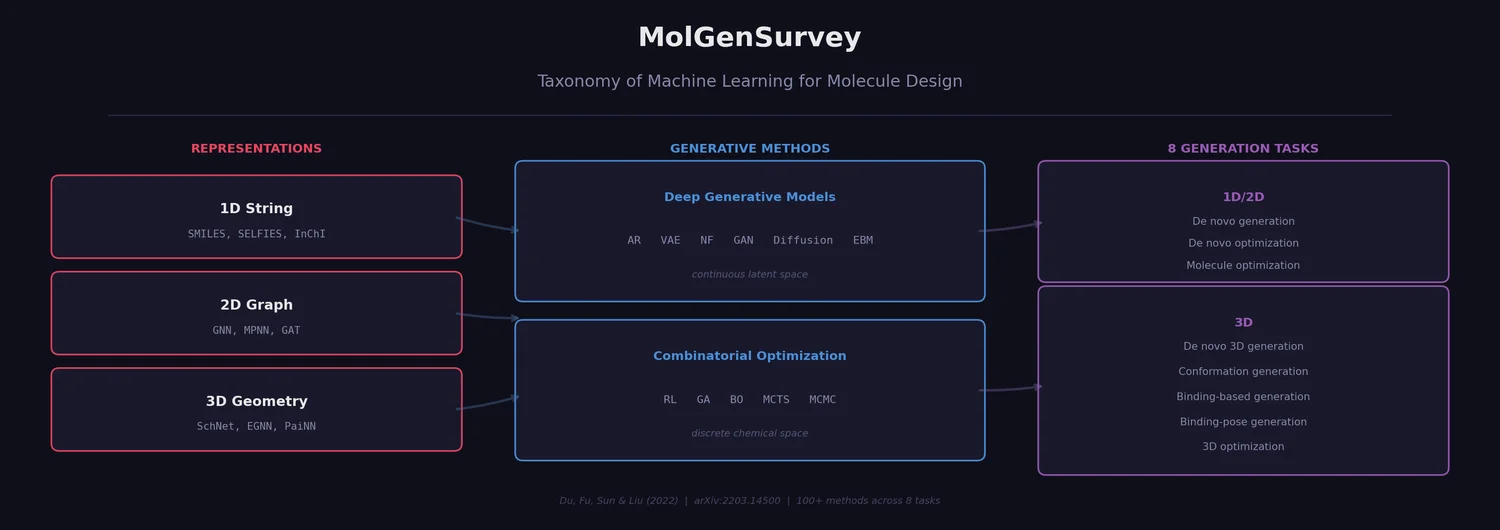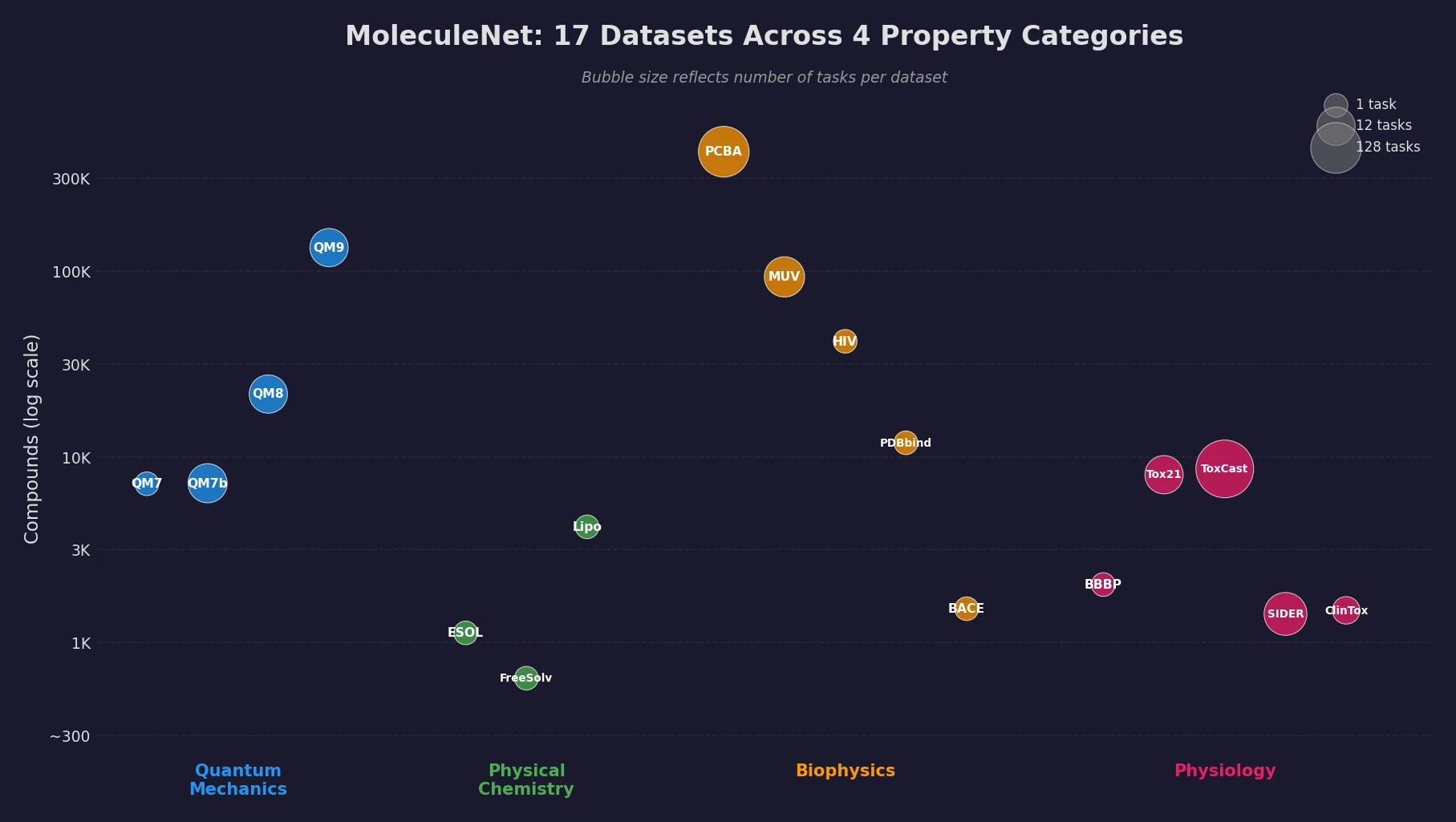
MoleculeNet: Benchmarking Molecular Machine Learning
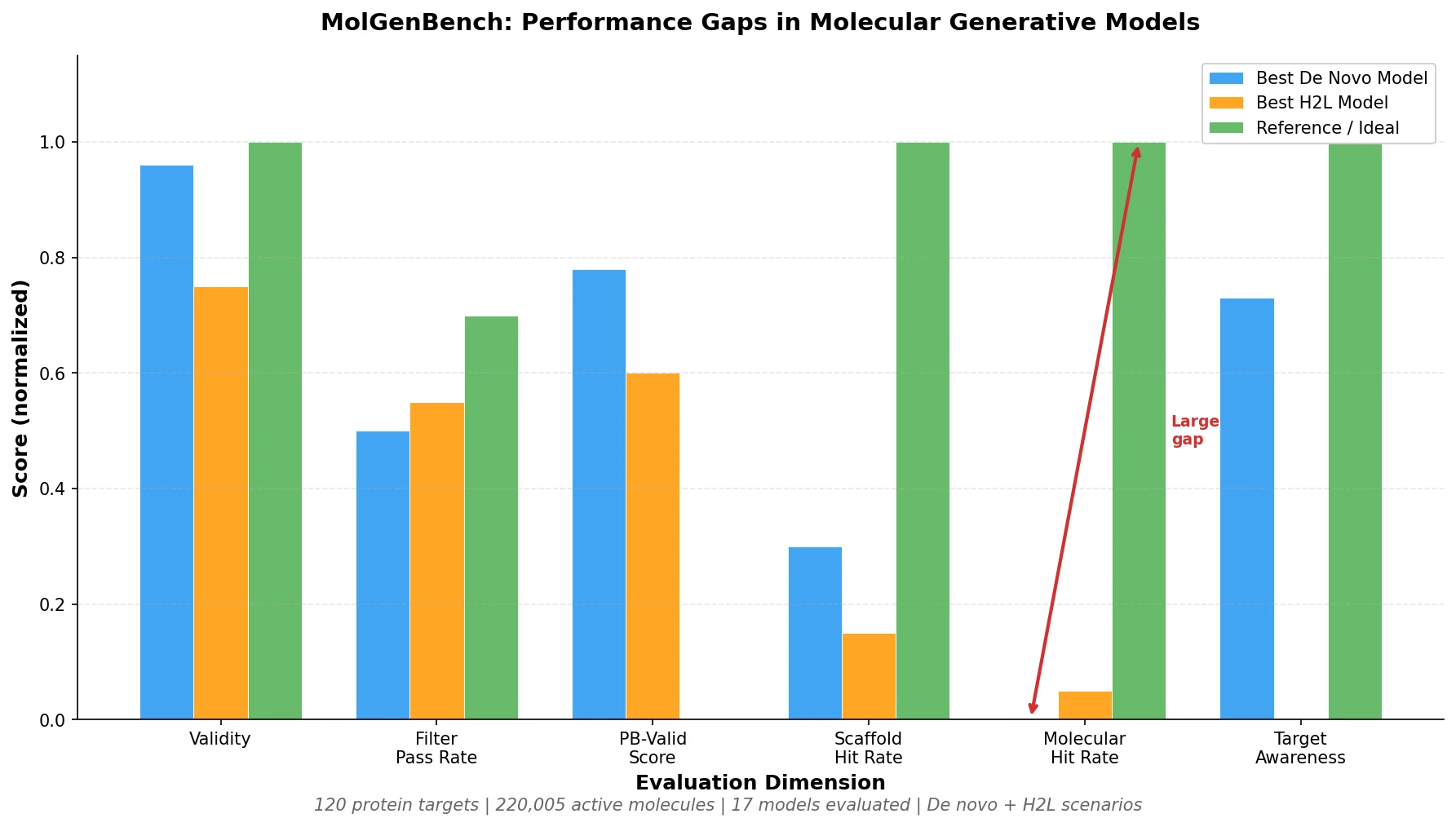
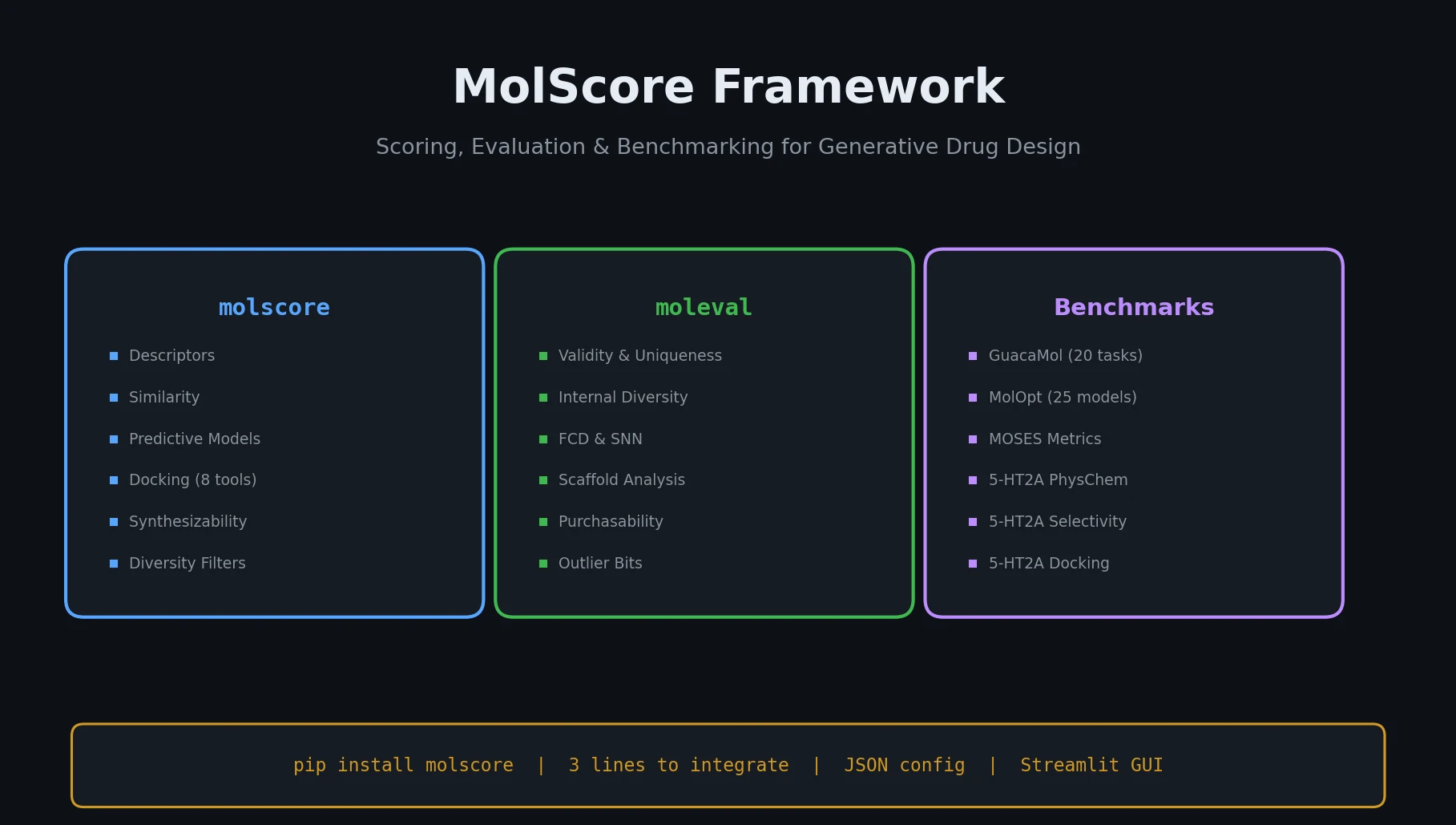
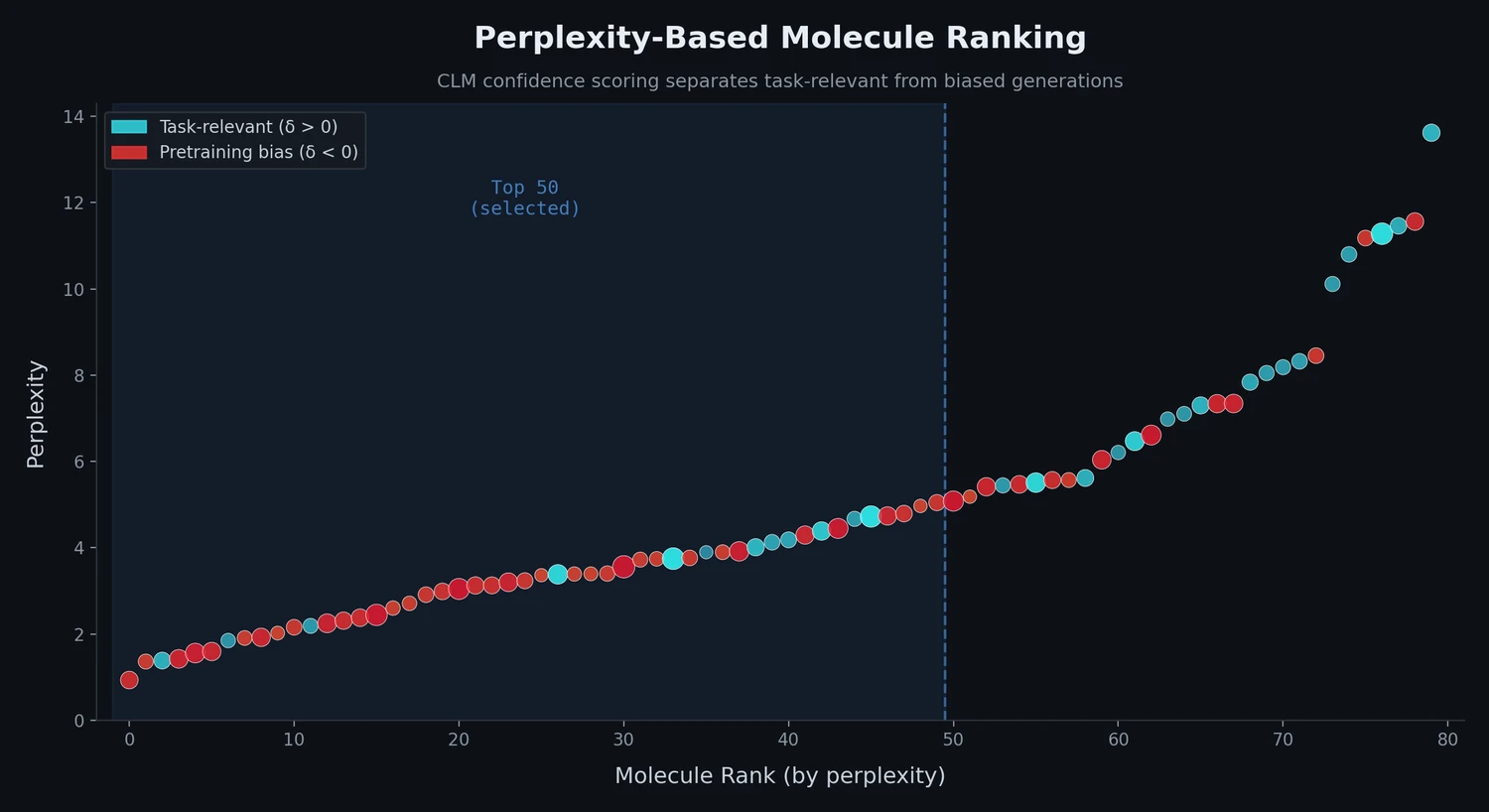
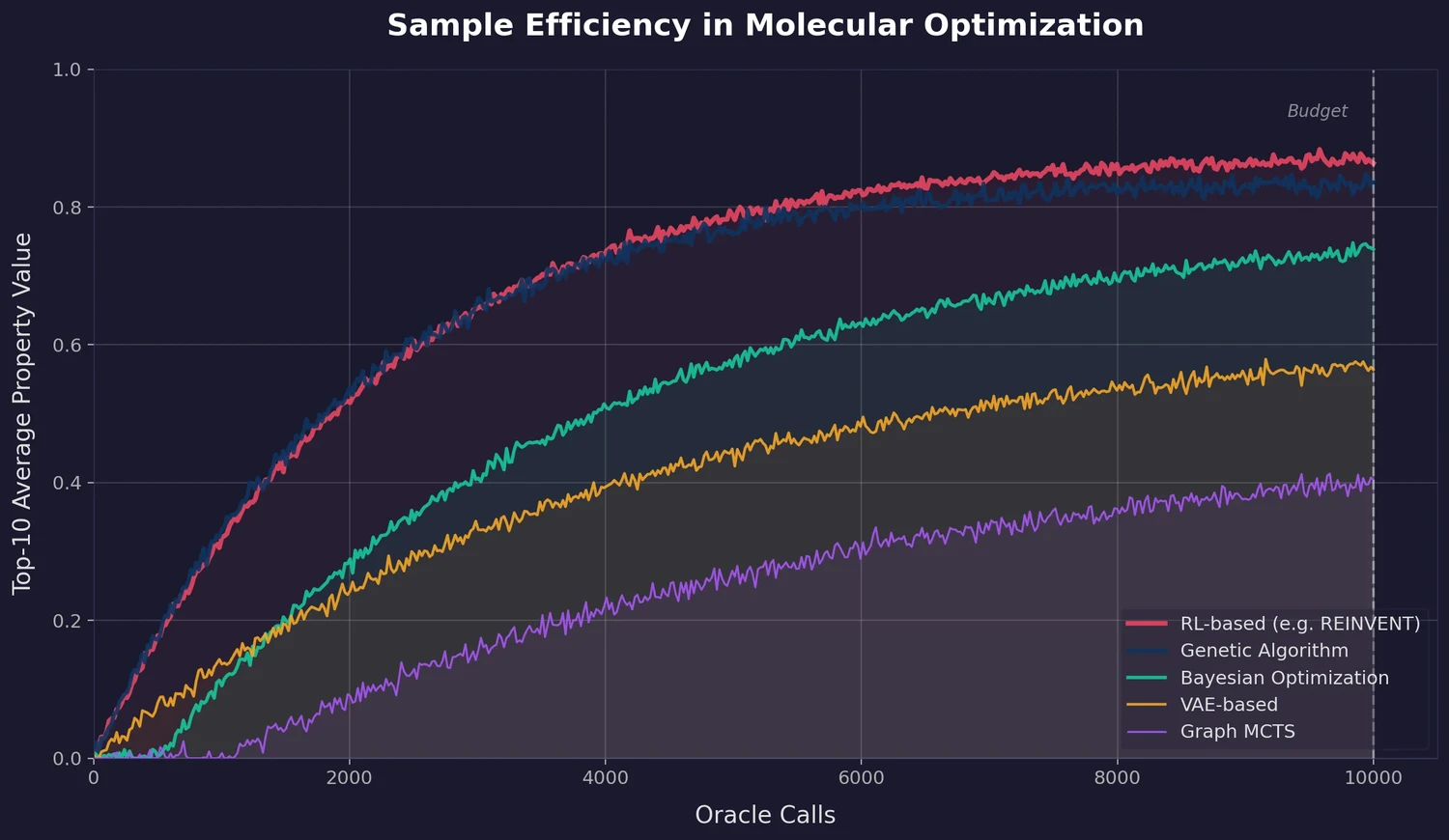
MoleculeNet introduces a large-scale benchmark suite for molecular machine learning, curating over 700,000 compounds across 17 datasets with standardized metrics, data splits, and featurization methods integrated into the DeepChem open-source library.