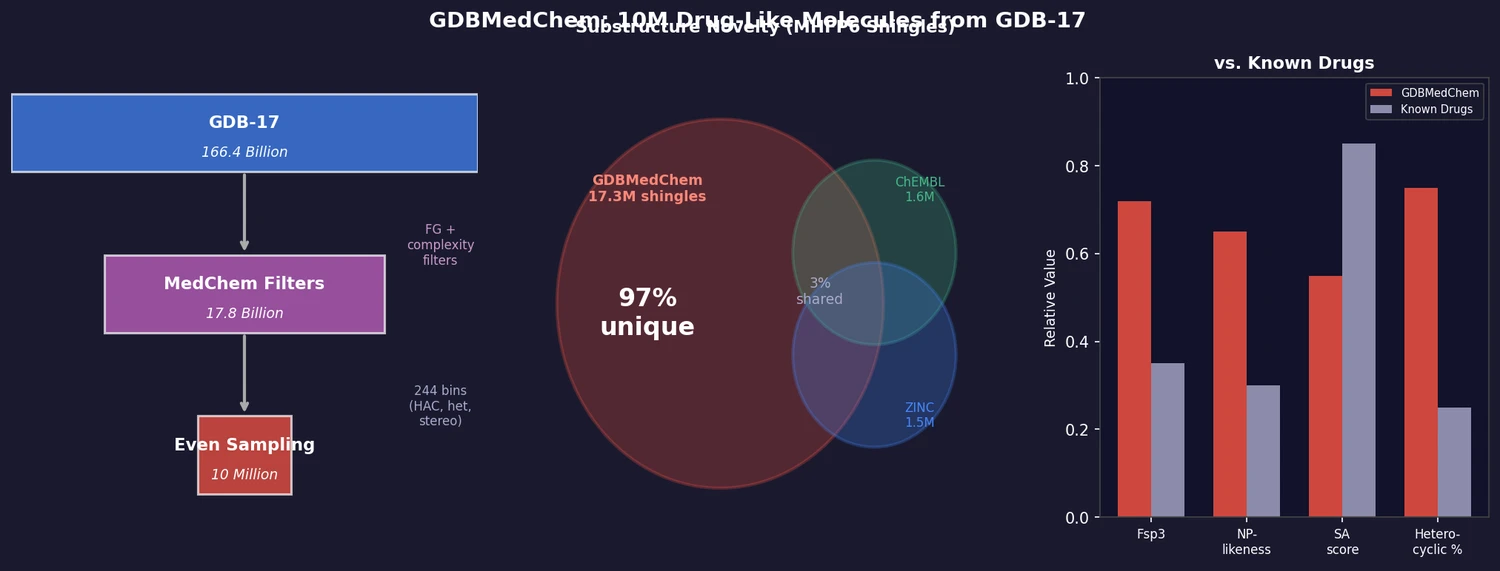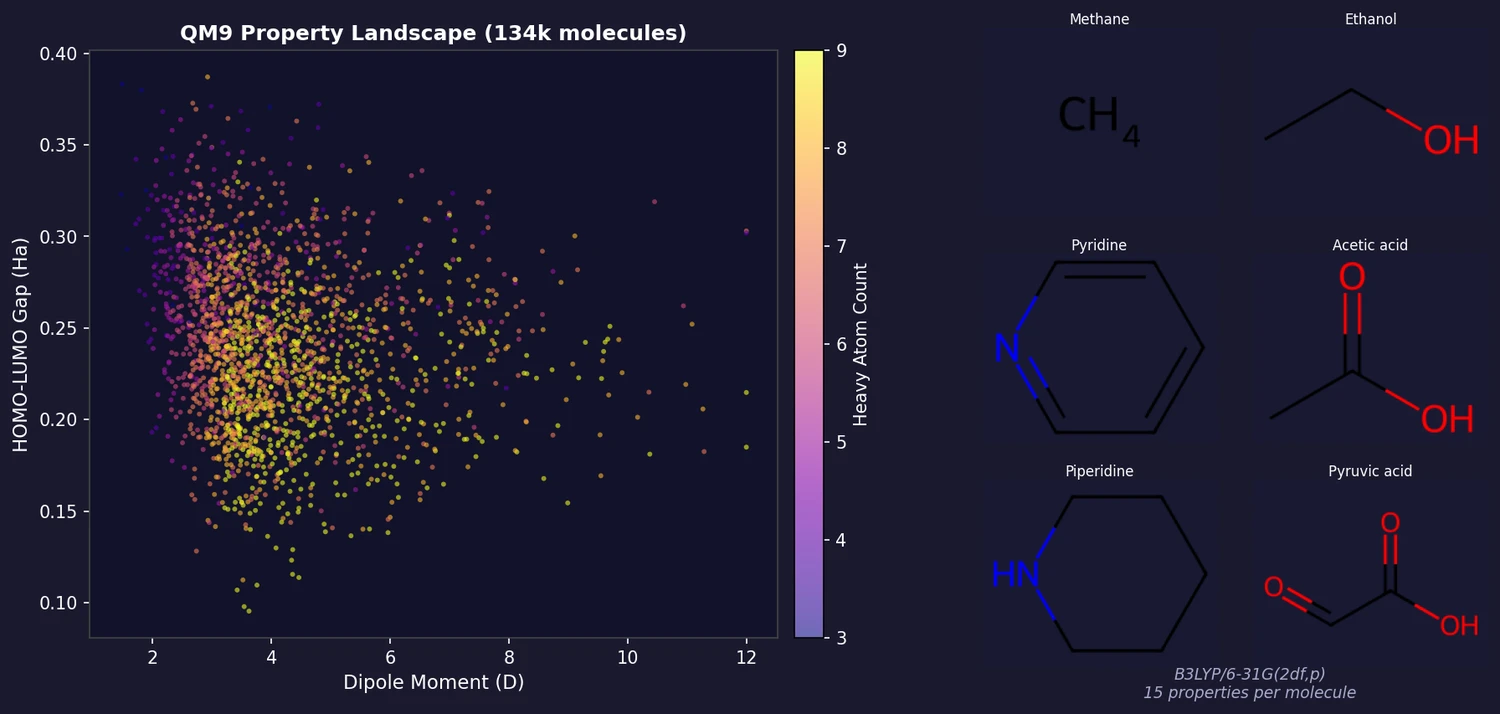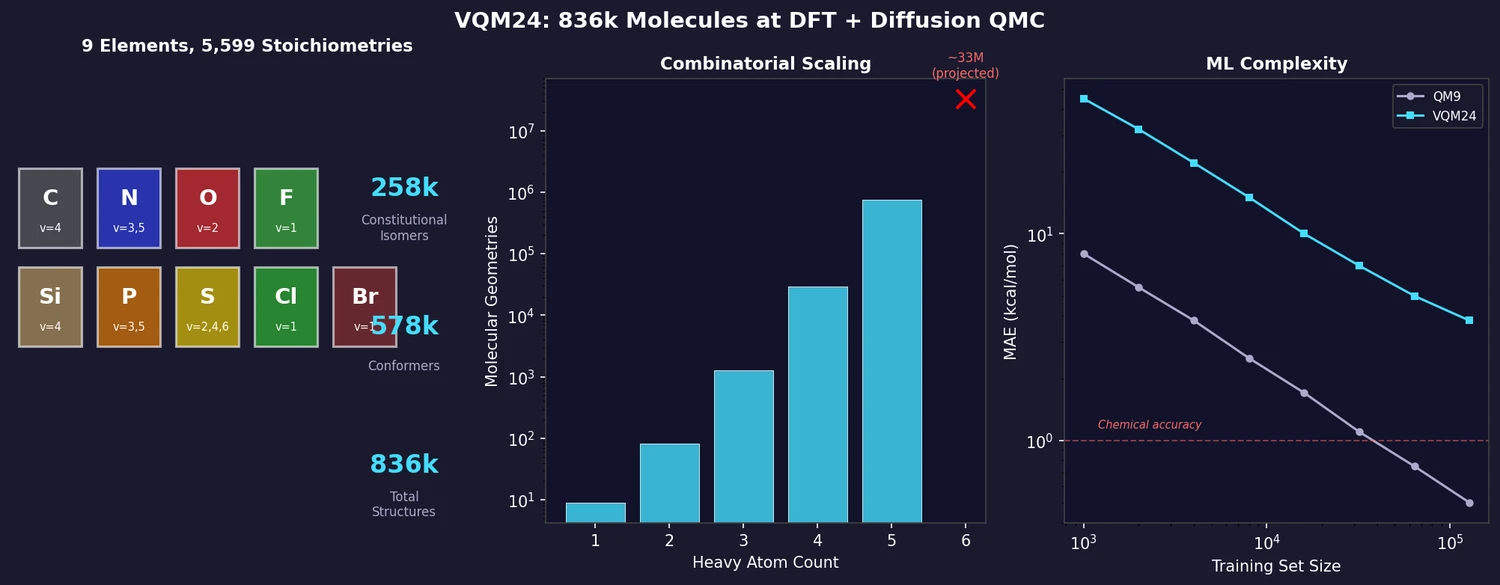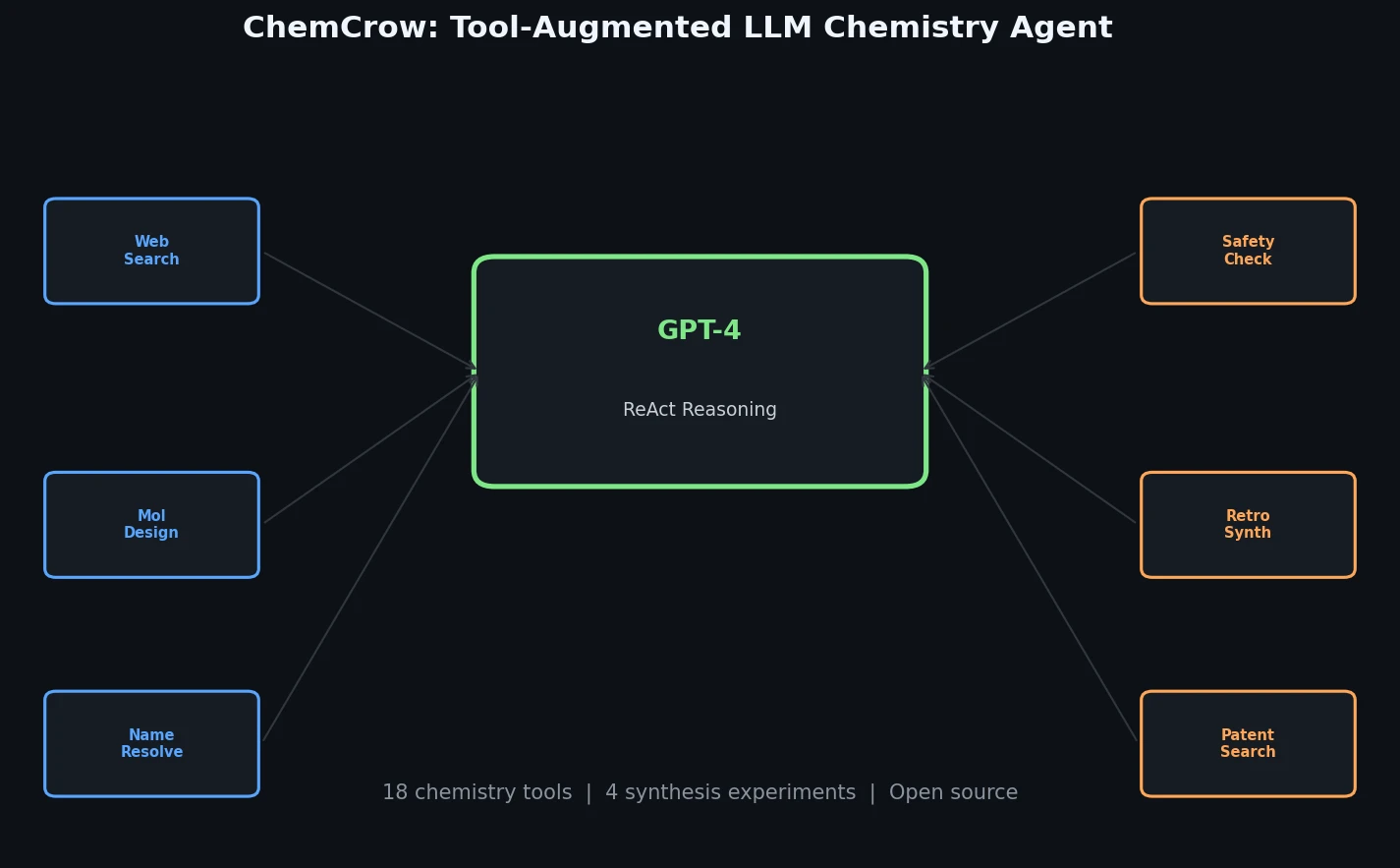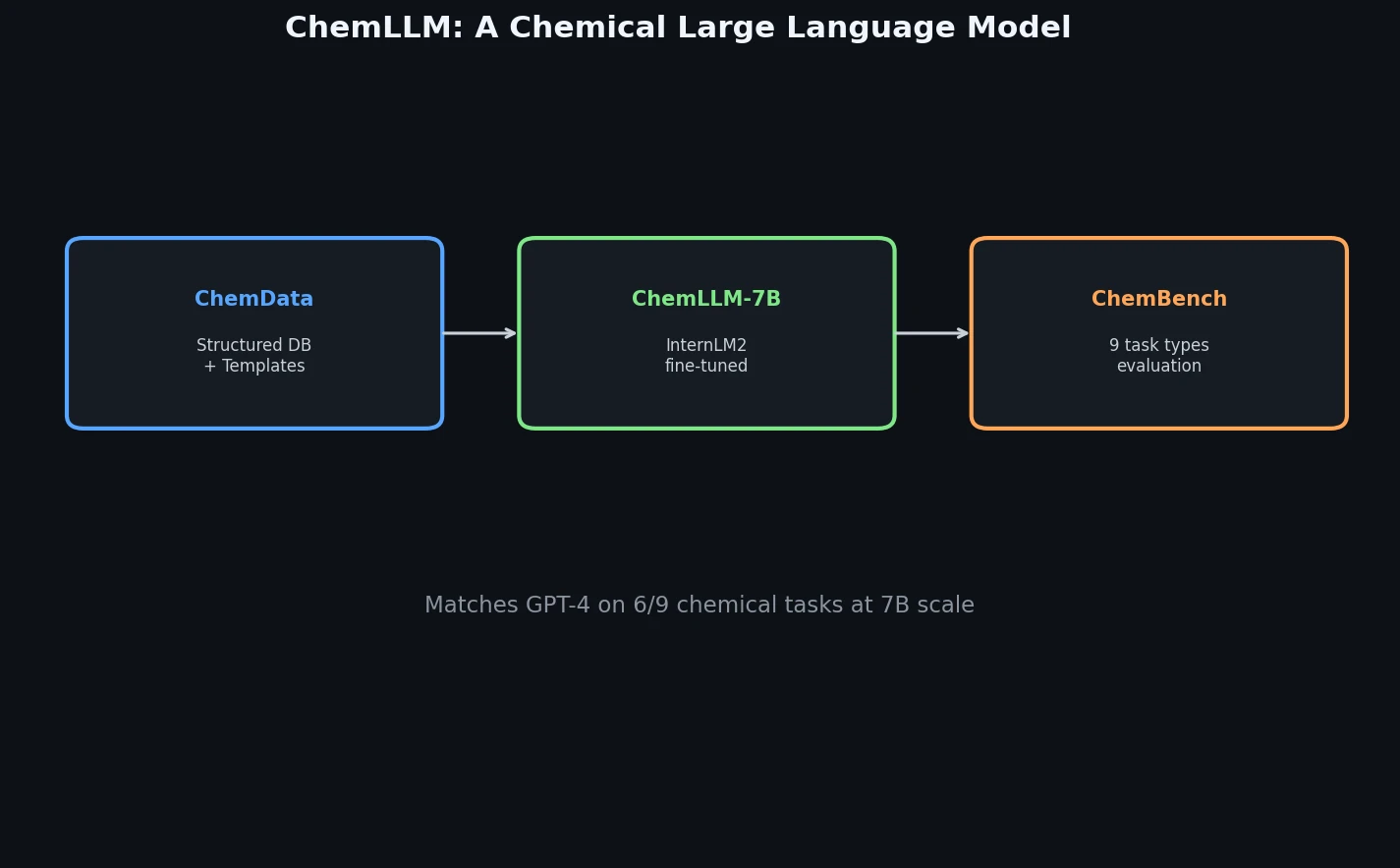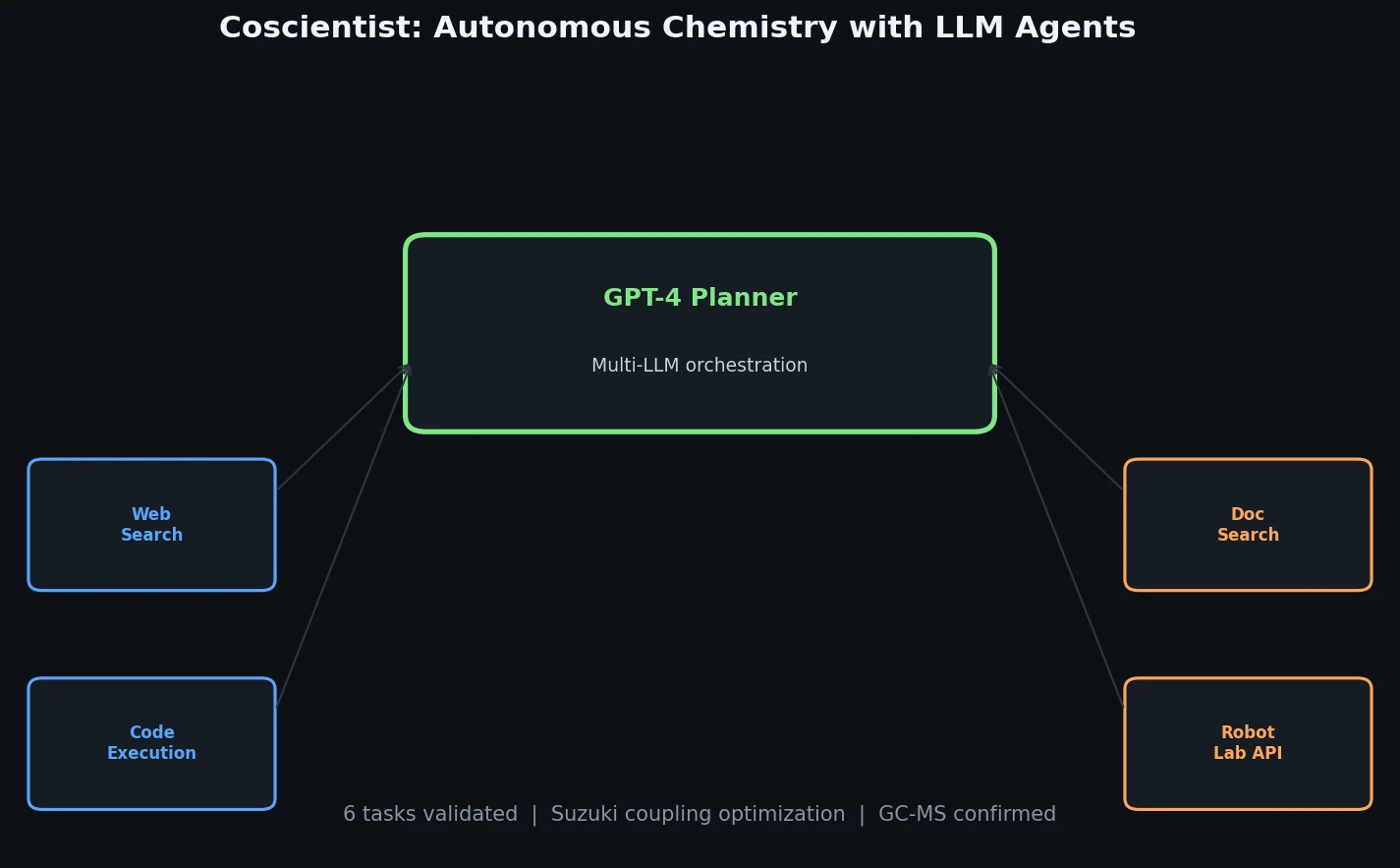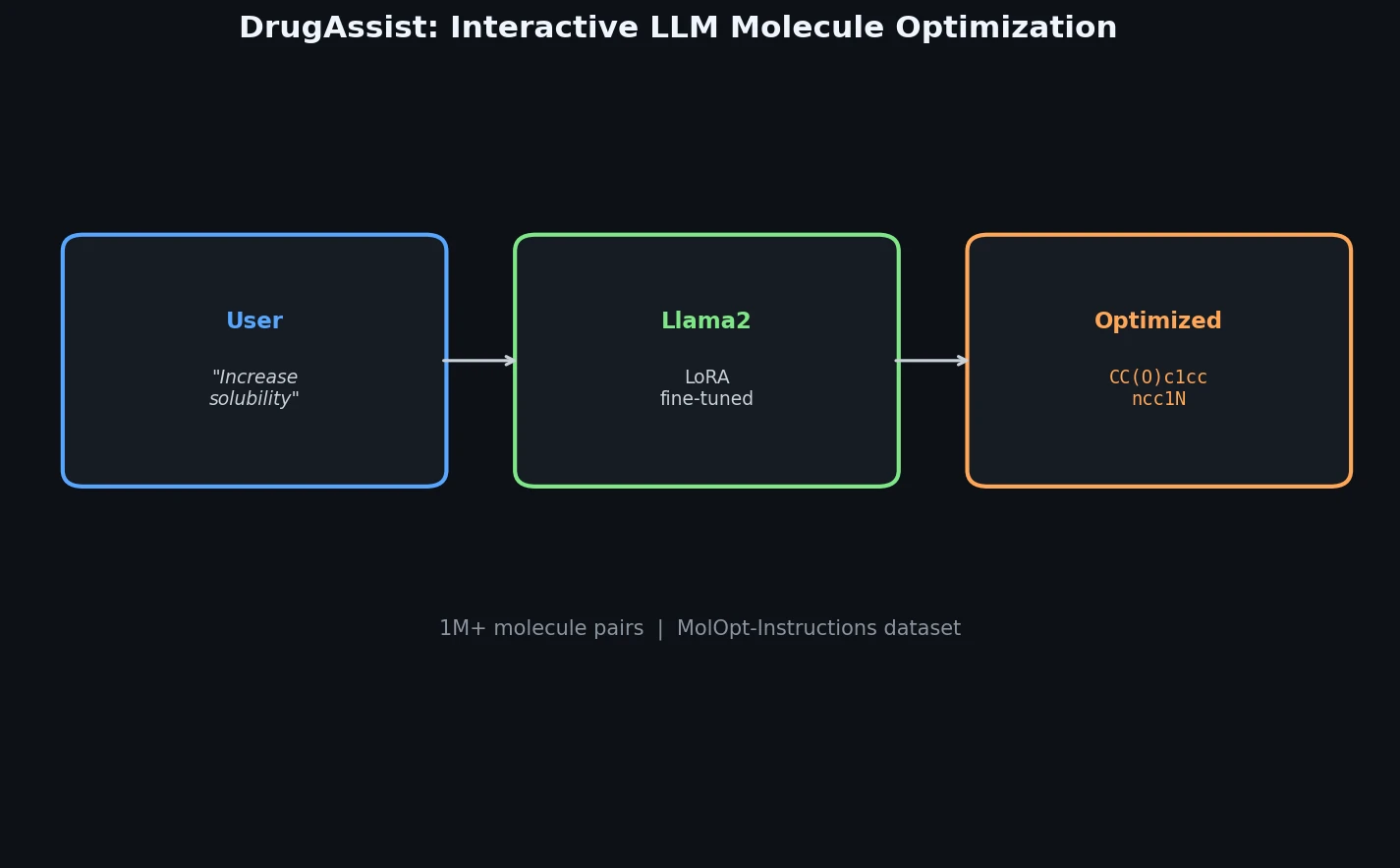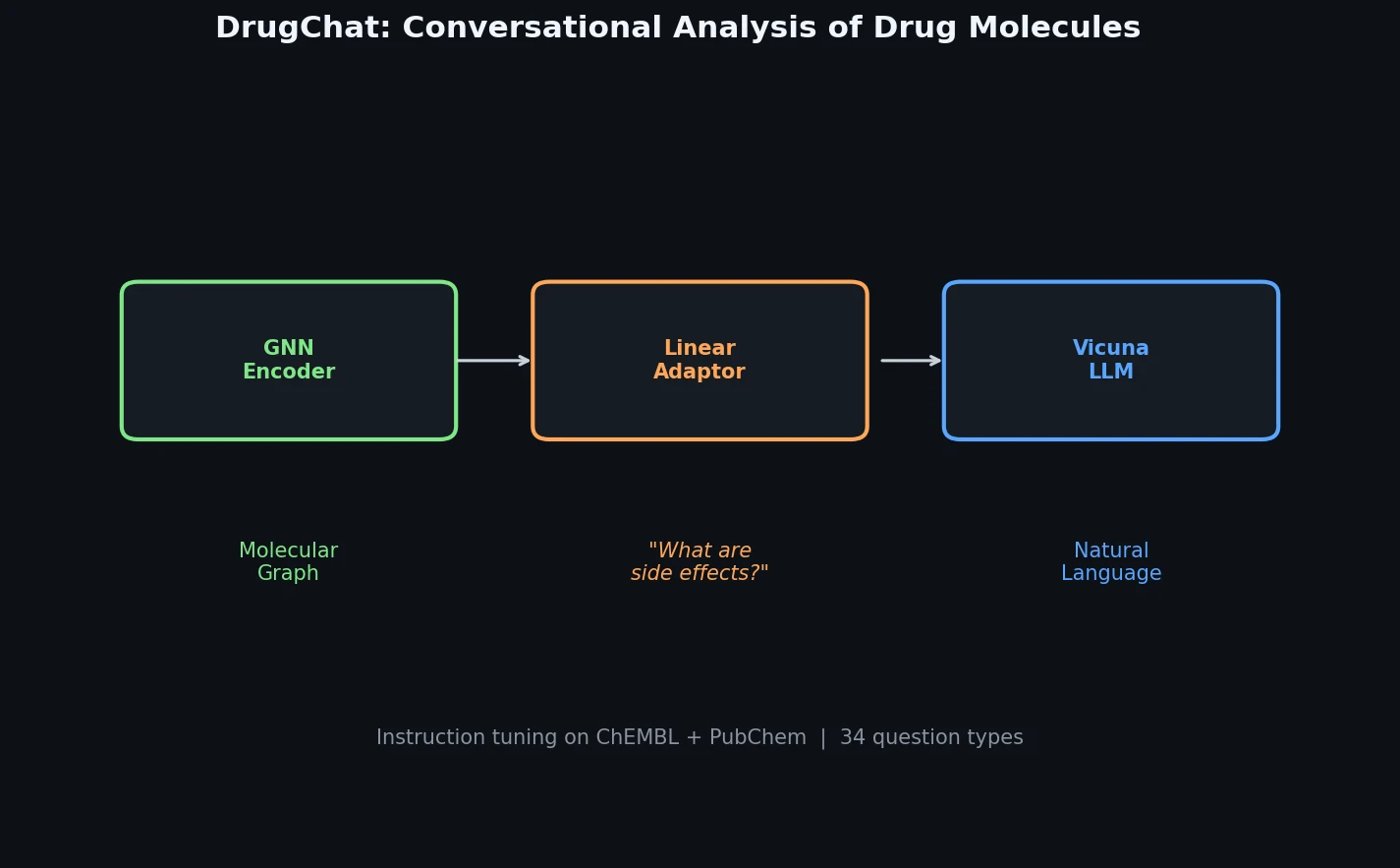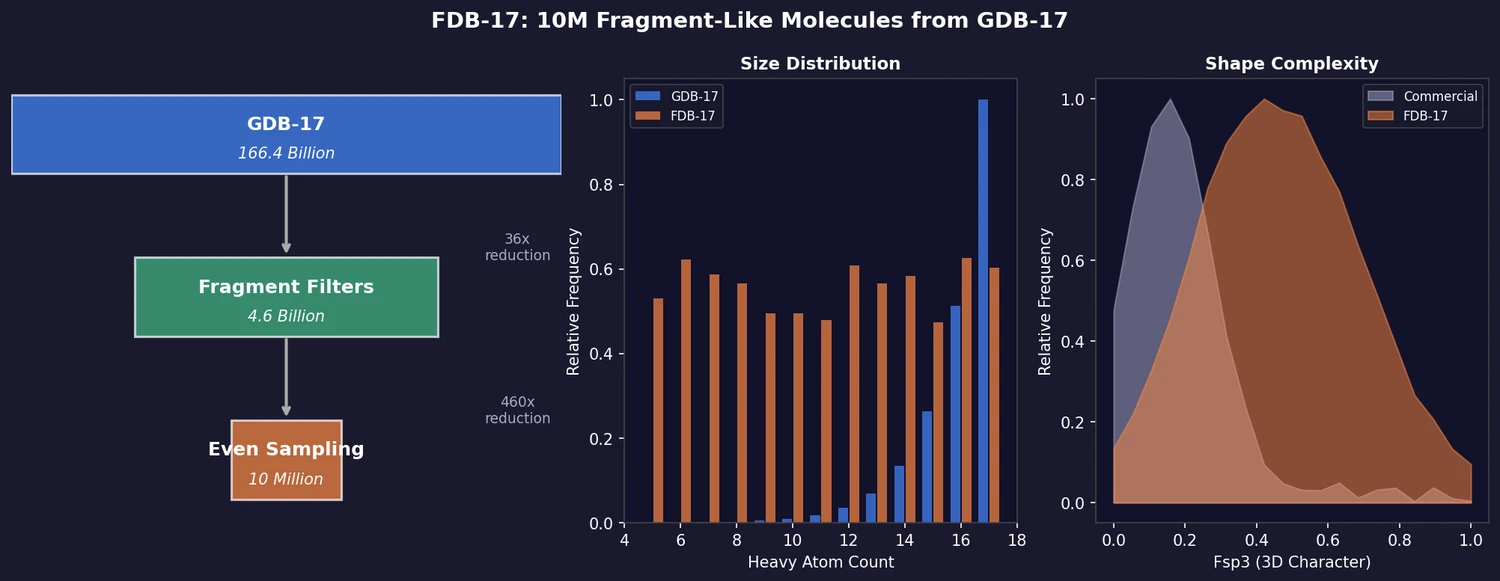
FDB-17: Fragment Database (10M Molecules)
FDB-17 contains 10 million fragment-like molecules selected from GDB-17’s 166.4 billion entries. Fragment-likeness filters reduce GDB-17 by 36x to 4.6 billion molecules, then even sampling across (HAC, heteroatoms, stereocenters) triplets produces a 460x further reduction to a manageable, diverse library enriched in 3D-shaped molecules.