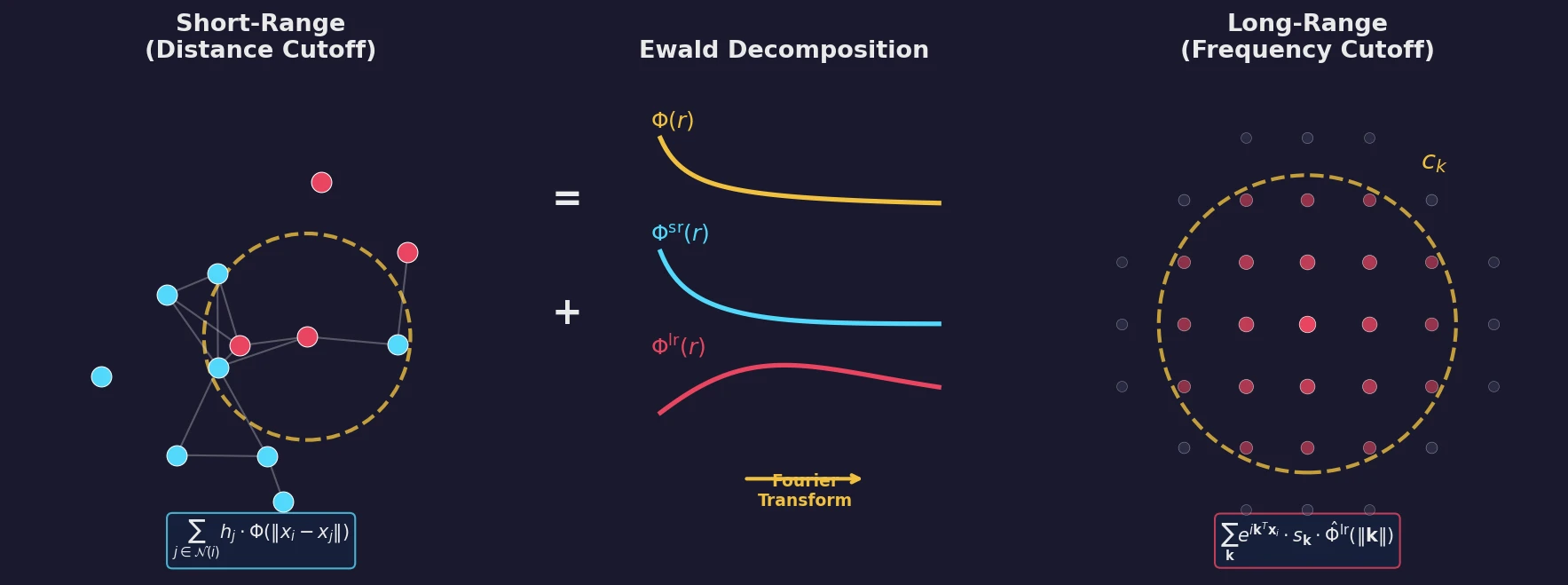
Ewald Message Passing for Molecular Graphs
Proposes Ewald message passing, a Fourier-space scheme inspired by Ewald summation that captures long-range interactions in molecular graphs. The method is architecture-agnostic and improves energy MAEs by 10% on OC20 and 16% on OE62 across four baseline GNN models.