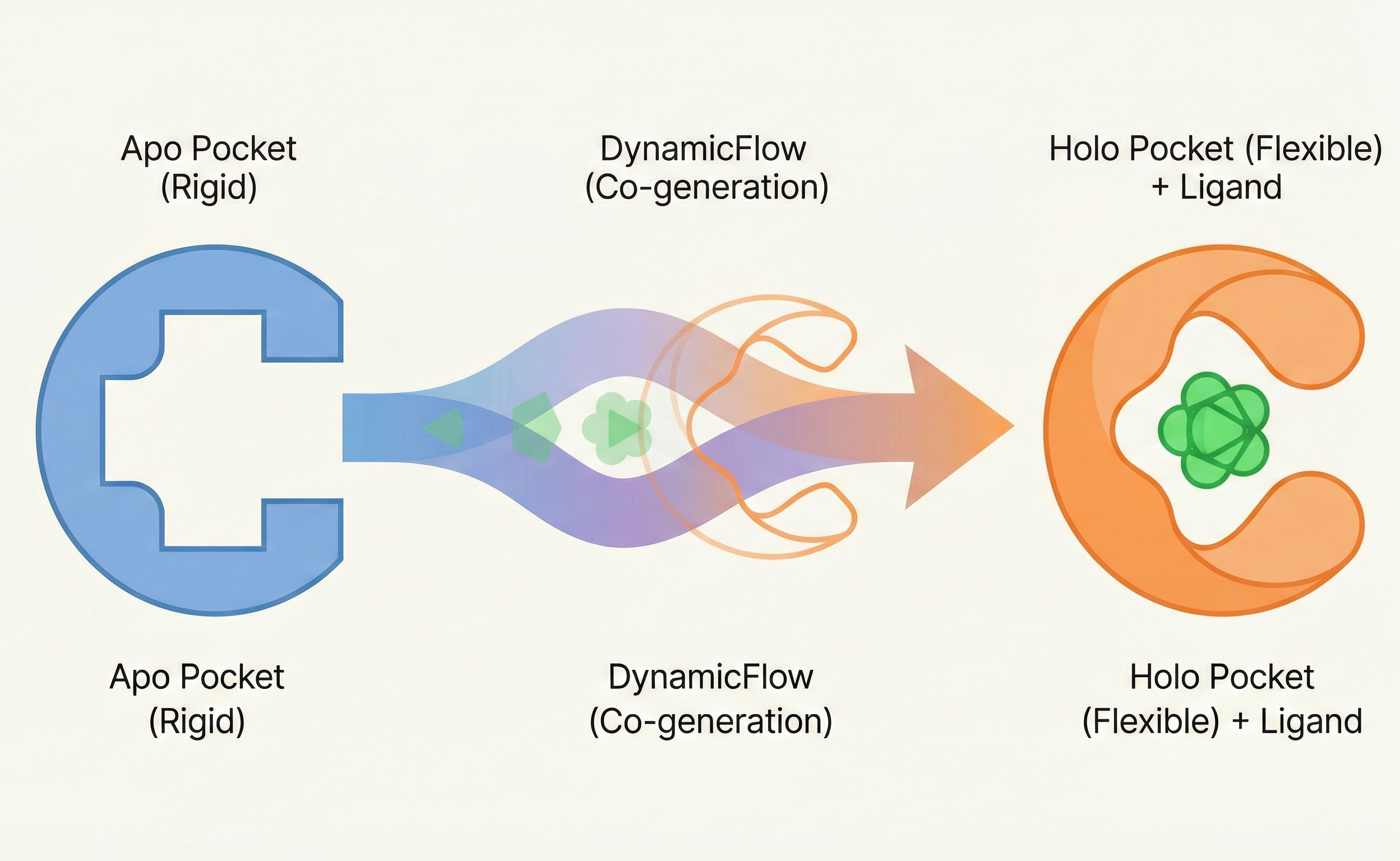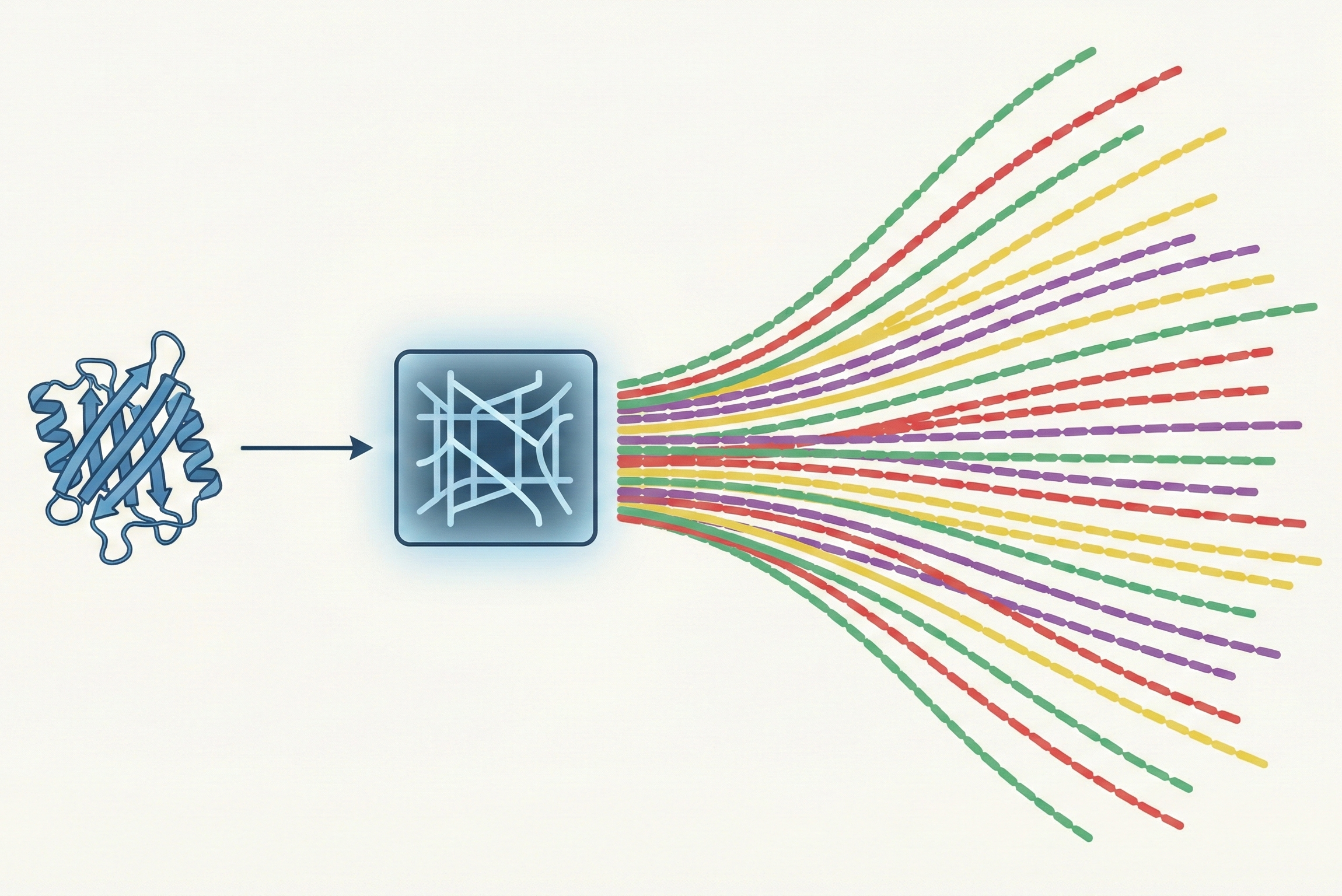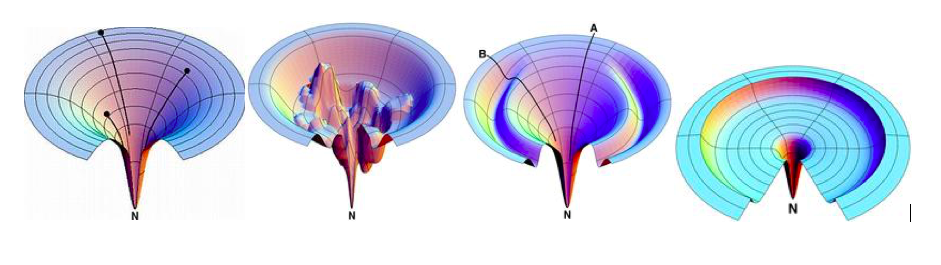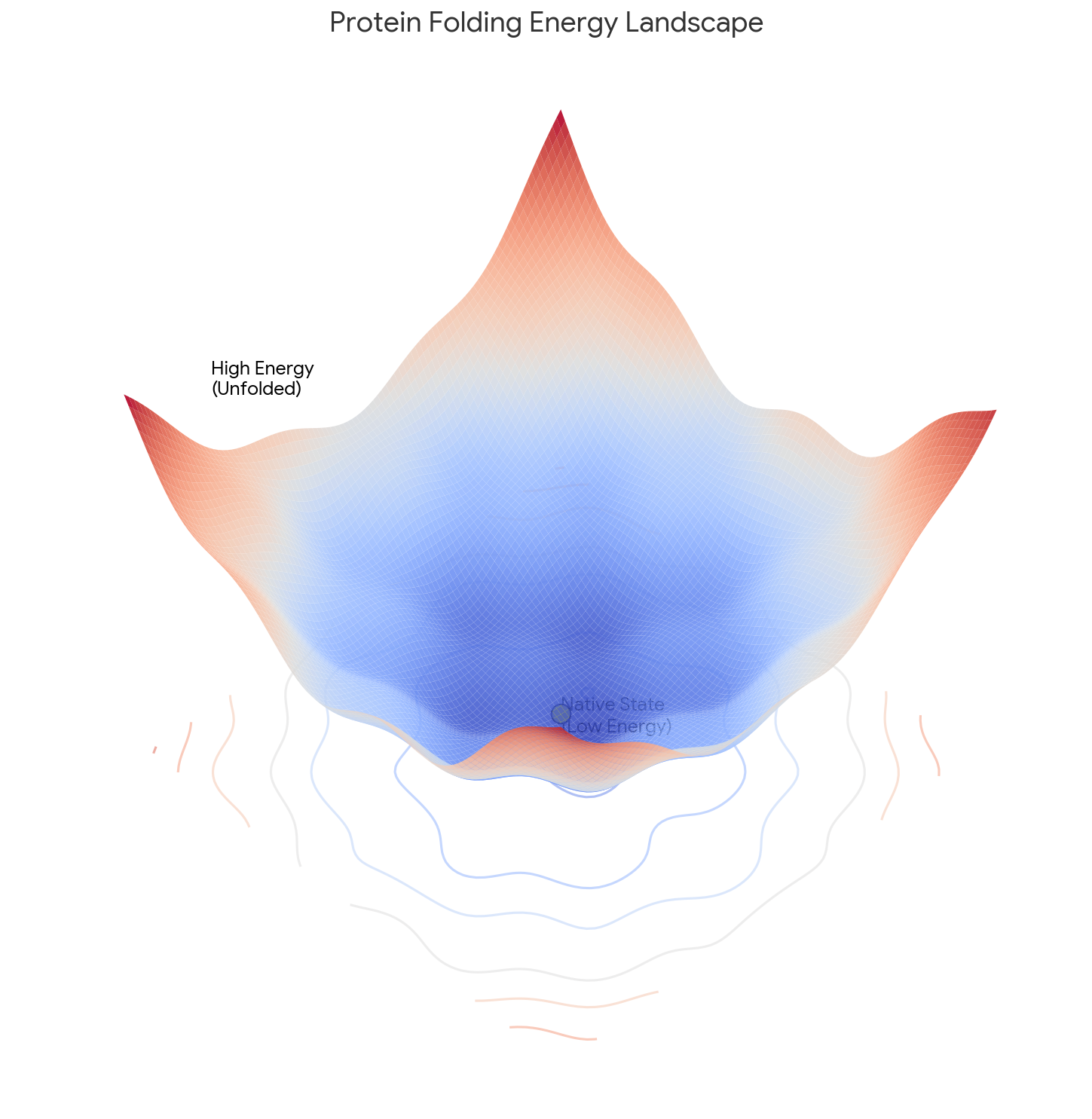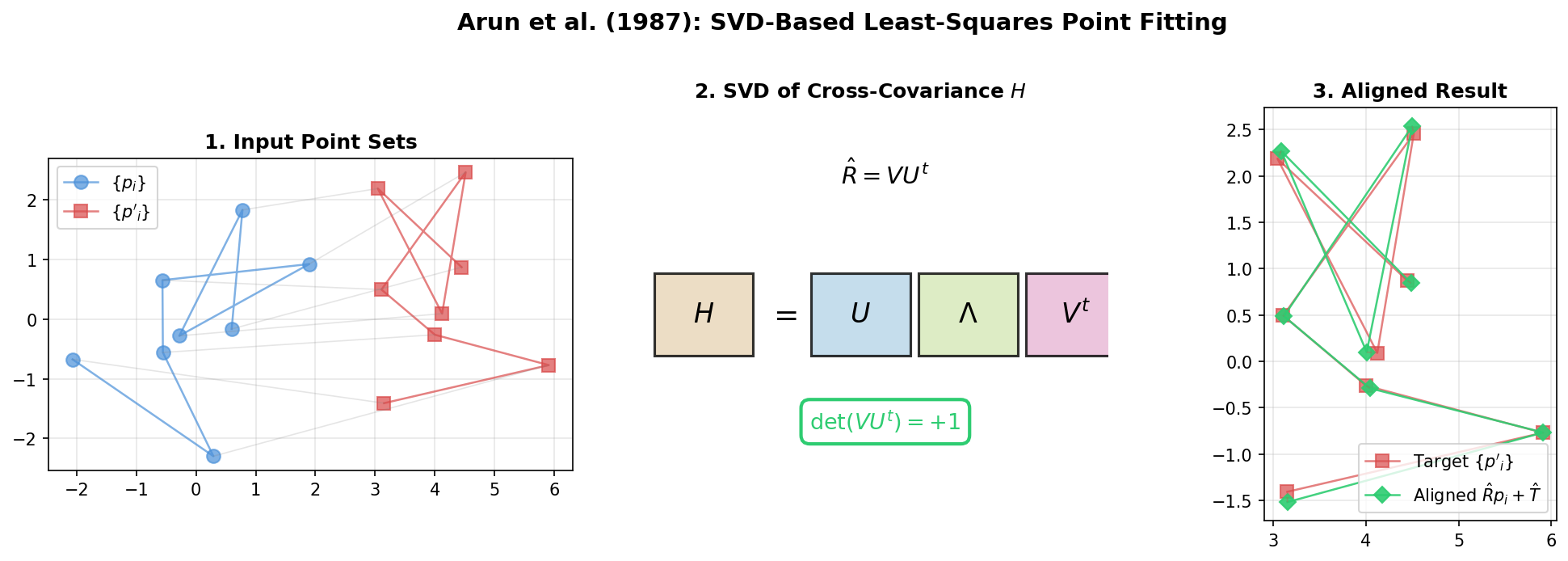
Arun et al.: SVD-Based Least-Squares Fitting of 3D Points
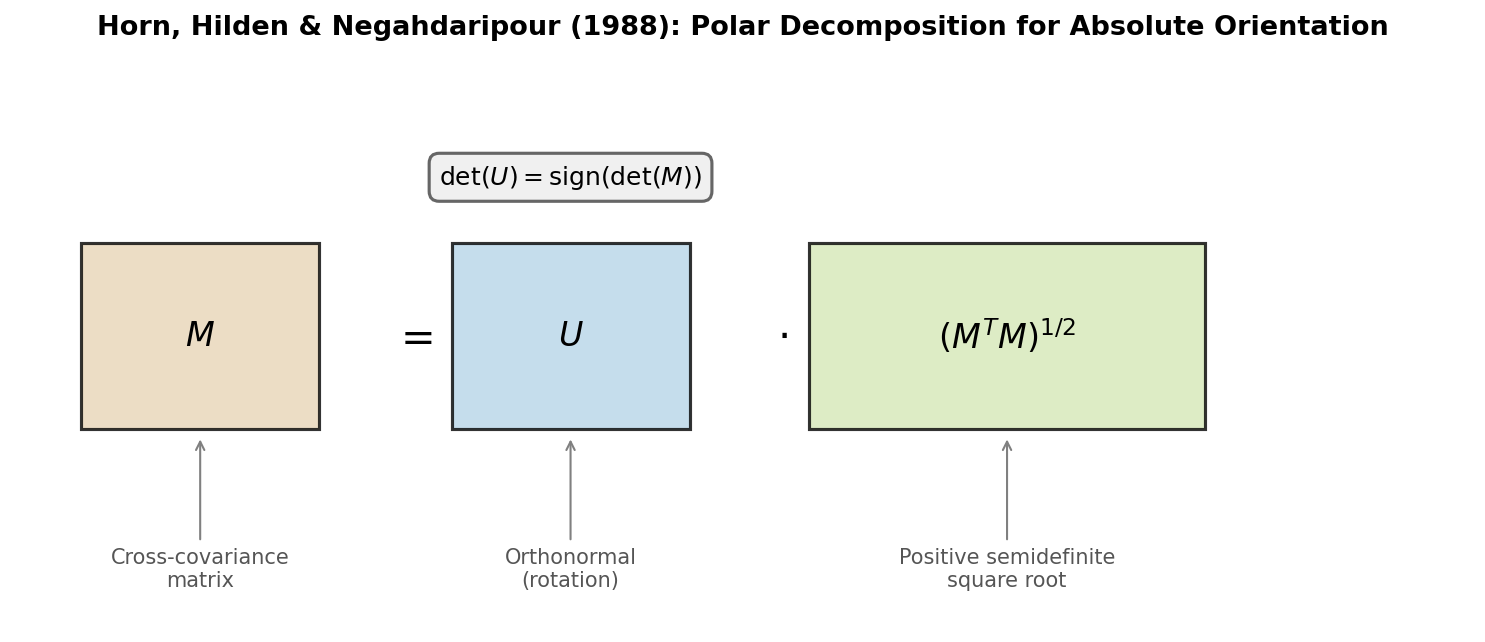
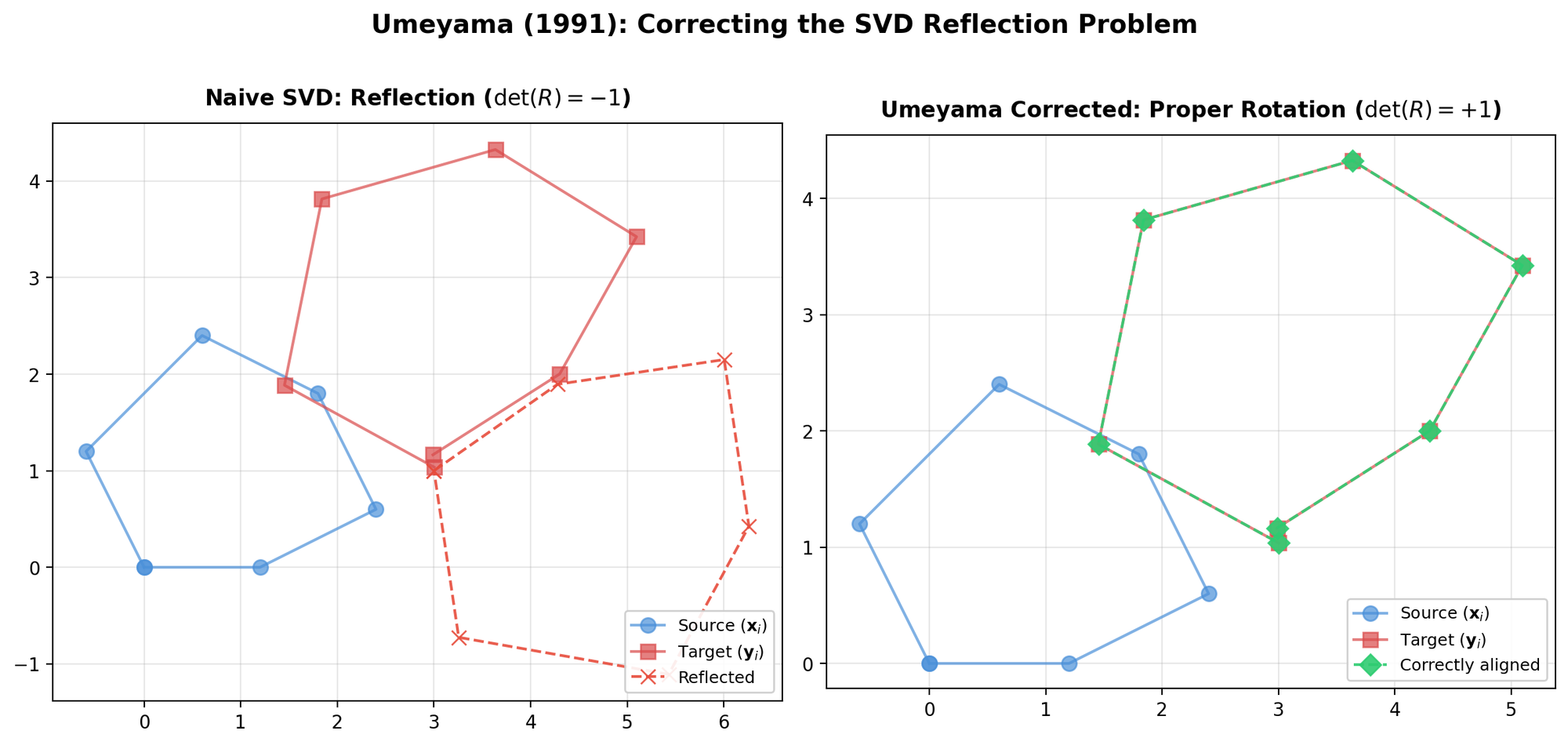
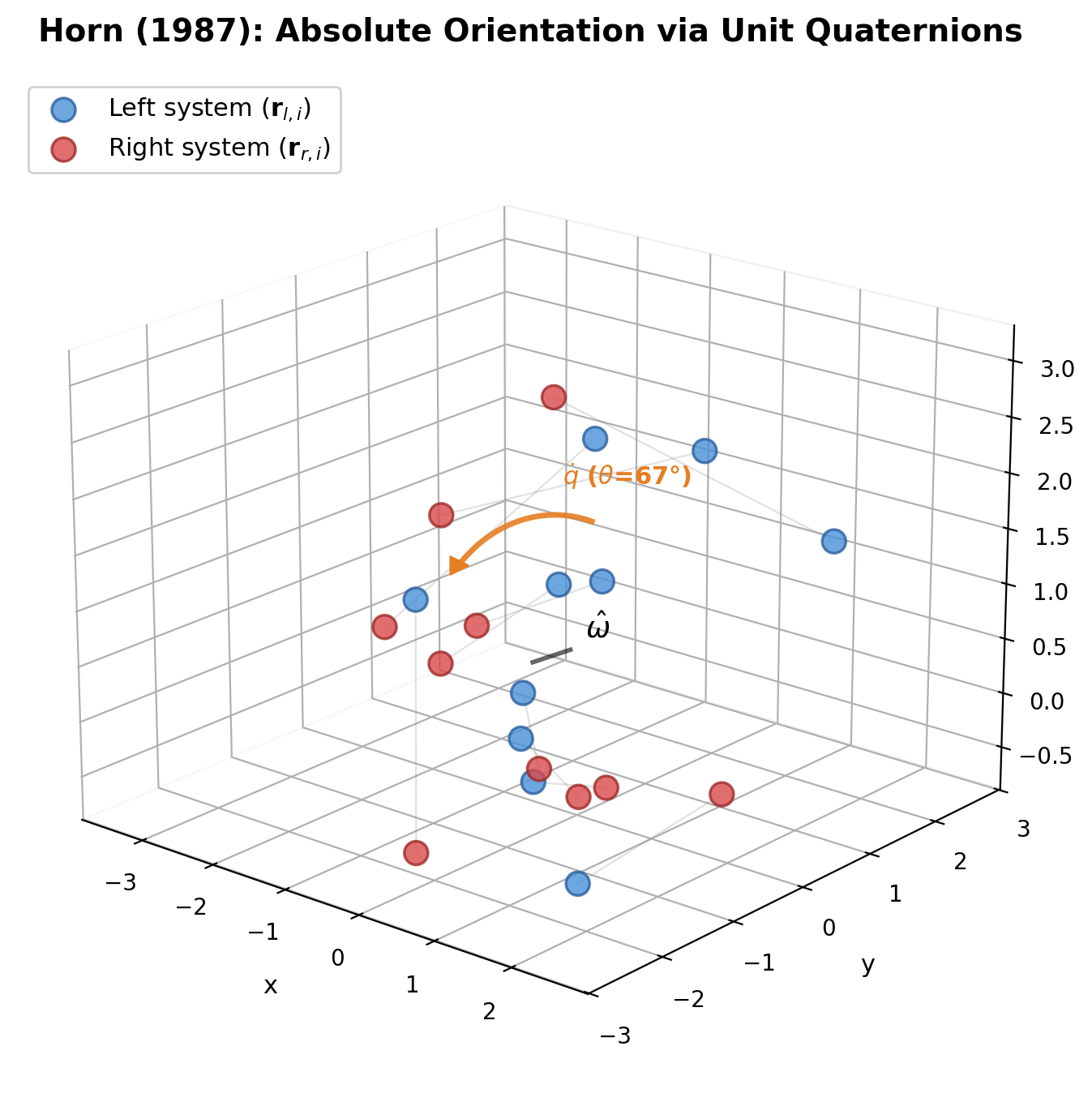
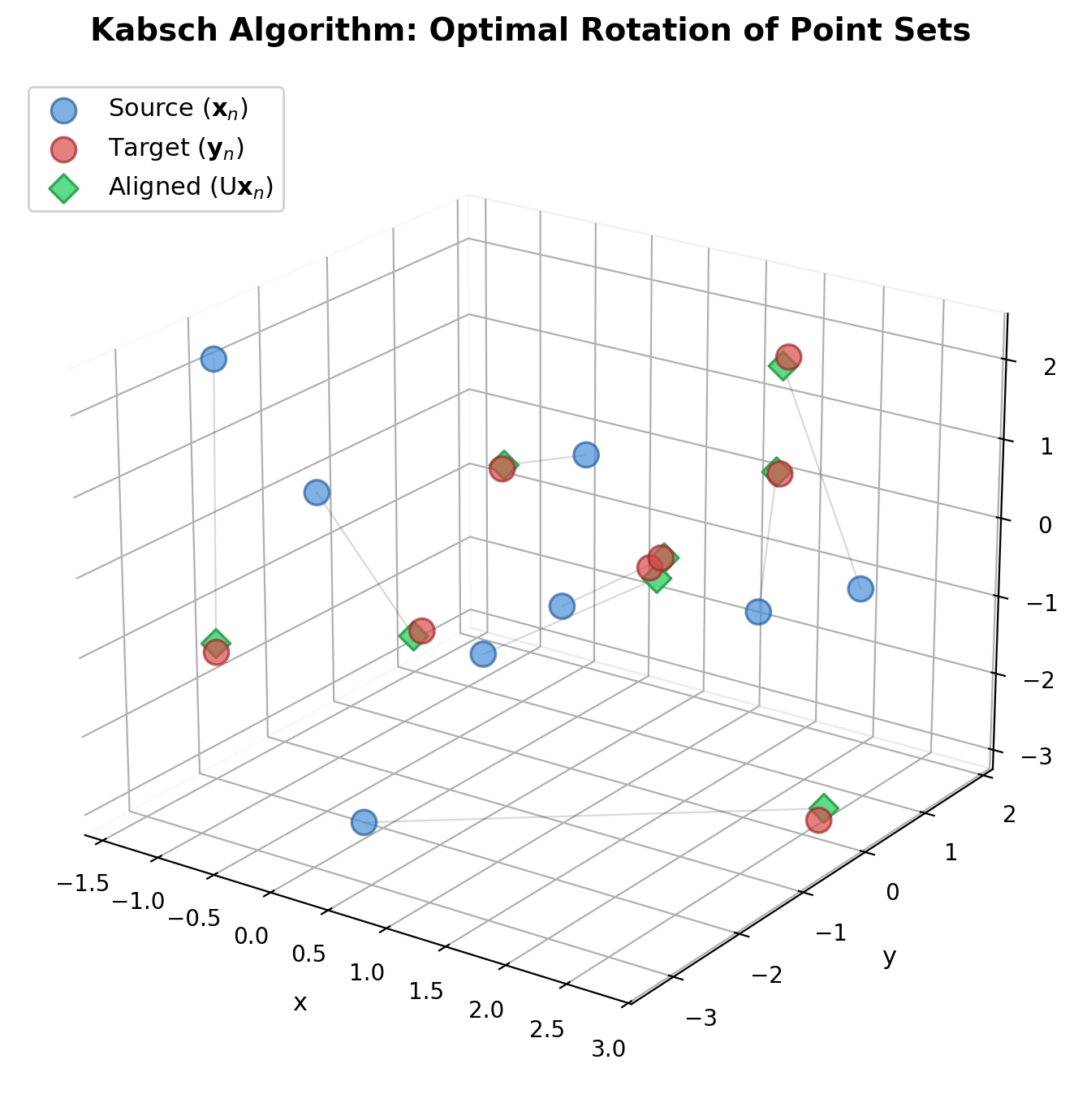
Presents a concise SVD-based algorithm for finding the optimal rotation and translation between two 3D point sets, with analysis of the degenerate reflection case that Umeyama later corrected.